
Mechanism and therapeutic strategy of hepatic TM6SF2-deficient non-alcoholic fatty liver diseases via in vivo and in vitro experiments
2022-07-11 08:30ZuYinLiGangWuChenQiuZhiJieZhouYuPengWangGuoHeSongChaoXiaoXinZhangGuiLongDengRuiTaoWangYuLongYangXiaoLiangWang
World Journal of Gastroenterology 2022年25期
Zu-Yin Li, Gang Wu, Chen Qiu, Zhi-Jie Zhou, Yu-Peng Wang, Guo-He Song, Chao Xiao, Xin Zhang, Gui-LongDeng,Rui-Tao Wang,Yu-Long Yang, Xiao-Liang Wang
Abstract
Key Words: TM6SF2; Nonalcoholic fatty liver disease; Fatty acid metabolism; Treatment; MK-4074
INTRODUCTION
The prevalence of nonalcoholic fatty liver disease (NAFLD) is rapidly increasing and is now more than one-quarter of the global population[1]. Approximately 6%–30% of patients with NAFLD evolve from simple steatosis (SS) to steatohepatitis, characterized by excessive hepatic lipid accumulation, inflammation, and hepatocyte damage[2,3]. Long-term steatohepatitis can cause liver fibrosis, and its adverse outcome may include liver dysfunction and hepatocellular carcinoma[4,5]. The prognosis of NAFLD and its related complications varies among individuals, given that genetic variants affect the occurrence and progression of NAFLD[6]. The development of genome-wide association studies and highthroughput technologies has allowed for in-depth analysis of genetic risk factors for NAFLD[7,8].Identification of genes with high-risk single nucleotide polymorphisms (SNPs) has allowed the screening of individuals with a genetic predisposition to NAFLD[9], and several studies have reported some potential SNP loci (e.g., PNPLA3 rs738409, MBOAT7 rs641738, and PPARGC1A rs2290602)associated with the susceptibility and progression of fatty liver[10-12]. To further elucidate the mechanistic basis of the link between potential SNPs and susceptibility to NAFLD, the functions of many candidate genes have been thoroughly investigated, includingTM6SF2.
TM6SF2 is a multi-transmembrane protein expressed predominantly in the intestine and liver,implying a metabolism-related function[13]. Our previous study and others have demonstrated that the rs58542926 locus of TM6SF2 confers susceptibility to NAFLD[14-16]. The nonsynonymous variant of TM6SF2, rs58542926 (E167K), leads to protein misfolding, acceleration of protein degradation, and,therefore, a reduction in TM6SF2 protein levels and gene function[15]. Recently, several studies have focused on the influence of TM6SF2 deficiency or its E167K mutant on cholesterol metabolism and its association with reduced very low-density lipoproteins (VLDL) content that causes a decrease in hepatic triglyceride (TG) output[17-19]. Considering that the liver is the central hub for lipid metabolism, there is a balance among the processes of uptake, synthesis, and intracellular expense of fatty acids under normal circumstances. Hepatic lipid accumulation may occur when this balance is disrupted[20].However, the regulatory role of TM6SF2 in fatty acid metabolism in the context of NAFLD remains largely unknown. Therefore, our study aimed to elucidate the influence of TM6SF2 on fatty acid metabolism in experimental NAFLD models.
The present study revealed that hepatic TM6SF2 expression was elevated in patients with NAFLD and mouse models. Our results suggest that TM6SF2 elevation is a compensatory response to NAFLD.Physiologically, the reactive overexpression of TM6SF2 can protect the liver and reduce hepatic lipid accumulation under the condition of a high-fat diet (HFD). Loss of TM6SF2 exacerbates hepatic lipid accumulation. Further investigation revealed that the enhanced processes of uptake and synthesis of fatty acids and impaired oxidation processes were observed in hepatic TM6SF2 deficiency, suggesting that TM6SF2 deficiency caused a pathogenic link between the metabolic dysfunction of fatty acids and hepatic steatosis in NAFLD. The elucidation of metabolic alterations suggested the way of therapeutic intervention, and our results showed that liver-specific acetyl-CoA carboxylase (ACC) inhibitor (MK-4074) would block the enhanced synthesis of fatty acid, improve fatty acid β-oxidation, and then reverse the NAFLD phenotypes caused by TM6SF2 deficiency. Collectively, this study suggested a protective role of TM6SF2 in HFD conditions, revealed a pivotal role of TM6SF2 in fatty acid metabolism, and suggested a therapeutic intervention for TM6SF2-deficient exacerbated NAFLD.
MATERIALS AND METHODS
Cell culture
Knockdown of theTM6SF2gene was mediated by the lentiviral vectors pLenti6.3-MCS-TM6SF2-EGFP(sh-TM6SF2) or pLenti6.3-MCS-EGFP (sh-Ctrl). Target sequence for sh-TM6SF2: TGACCTGGCCCTTGTCATATA. After two or three generations of antibiotic screening, the expression of TM6SF2 was evaluated by immunoblotting. All cells were cultured in DMEM containing 10% fetal bovine serum(FBS) and 1% penicillin-streptomycin (Cat. No. C125C5, NCM Biotech, Suzhou, China) at 37 ℃ and 5%CO2. The cells were cultured under 1% O2and 5% CO2for 24 h to simulate hypoxic conditions. For starvation, cells were cultured in DMEM containing 0.1% FBS for 24 h.
Western blot
Cells in a 6-cm dish at 80% confluence were lysed with 250 μL of RIPA lysis buffer and centrifuged at 13000 rpm. The quantification of the supernatant collected was assessed using the BCA method(Beyotime, Jiangsu, China). Proteins were transferred to a 0.45 μm PVDF membrane according to the standard immunoblotting protocol after electrophoresis. The membrane was blocked in 5% fat-free milk and then maintained in primary antibodies for 12 h at 4 ℃. After washing, the membrane was incubated with HRP-labeled goat anti-rabbit (mouse) antibody at 25 ℃ for 60 min and visualized using an ECL reagent (Millipore, MA, United States). Supplementary Table 1 lists the antibodies used in this study.
Clinical specimens
Liver specimens were collected from the Shanghai General Hospital. Parts of the specimens were snap frozen in liquid nitrogen and then transferred to a -80℃ refrigerator to detect mRNA levels, and some were stored in formaldehyde solution for later pathological examination. When ≥ 5% of hepatocytes had steatosis, NAFLD diagnosis was confirmed. The diagnosis of the tissue sections was independently reviewed by two pathologists. This study was approved by the Ethics Committee of the Shanghai General Hospital and was performed strictly in accordance with the Declaration of Helsinki. All patients or their family members were fully informed of the study and signed written consent forms.
Immunohistochemistry
Paraffin-embedded liver tissues were deparaffinized using standard protocols and rehydrated. After antigen retrieval, the tissue was blocked and incubated with an anti-TM6SF2 (1:200) antibody overnight at 4 ℃. Then the slides were washed and incubated with biotin-labeled goat anti-mouse IgG (H + L) at 37 ℃ for 15 min and developed with a DAB work solution. Images were obtained using a microscope(Leica, Germany).
Real-time polymerase chain reaction assay
A total of 50 mg of liver samples from mice or humans was lysed with RNAiso Plus reagent (Takara Biotechnology, Otsu, Japan), and total RNA was extracted according to the standard protocol. Reverse transcription of 500ng of RNA was performed using the RNA polymerase chain reaction (PCR) Kit(Takara Biotechnology), and the resulting cDNA was used as the PCR template. Quantification of target gene expression was performed using a LightCycler®96 (Roche, Switzerland) and a Hieff®quantitative PCR SYBR Green Master Mix (No Rox) kit (Yeasen Biotechnology Co., Ltd, China). The mRNA level ofβ-actinwas used as the endogenous control. We chose the TM6SF2 level of one patient as the reference,and the TM6SF2 levels of all other patients were presented several times of this reference. The primers used are listed in Supplementary Table 1.
Experimental animals
The AAV system (type 8) expressing TM6SF2 short hairpin RNA (shRNA) (AAV-shTm6sf2) or overexpressing human TM6SF2 (AAV8-TM6SF2) was used to regulate the levels of TM6SF2 in C57BL/6 mice.The corresponding controls were AAV-shNC and AAV8-vector, respectively. All mice were fed a normal chow diet (NCD) for three days, and then 100 μL of AAV8 virus (2 × 1011) was injected into the tail vein. Transfection efficiency was determined by immunoblotting. Male mice weighing 19–22 g (aged 4–6 wk) were housed in a 23 ± 2°C environment with a 12 h light/dark cycle. All mice were allowed water ad libitum and were continuously fed an HFD or NCD for 16 wk. For MK-4074 treatment, the mice were primarily fed an 8-wk HFD to induce NAFLD phenotypes and then administered MK4074(10 mg/kg/day) or placebo (normal saline) for an additional eight weeks on the same diets.
Evaluation of serum parameters and hepatic lipid content
The concentrations of cytokines, metabolites, and hepatic enzymes in serum were determined using commercial kits [ab208348 for tumor necrosis factor (TNF), ab197742 for interleukin (IL)-1β, ab222503 for IL-6, JLC049 for CCL2, JLC5800 for CXCL10, ab180875 for acetoacetate, ab180876 for β-hydroxybutyrate (β-OH), C010-2-1 for aspartate transaminase (AST), and C009-2-1 for alanine aminotransferase(ALT)] according to the manufacturer’s instructions. The hepatic lipid content and malonyl-CoA levels were also measured using commercial kits [A110-1-1 for TG, A111-1-1 for total cholesterol (TC), A042-2-1 for non-esterified fatty acids (NEFA), and JL47416 for malonyl-CoA], and normalized by the total protein. The details of all the commercial kits used are listed in Supplementary Table 1.
Microarrays
After palmitic acid (PA, 150 μmol/L) stimulation for 24 h, total RNA was extracted from both TM6SF2-knockdown L02 cells and the corresponding controls using RNAiso Plus (Takara Biotechnology).Microarray gene expression analysis was performed by Illumina sequencing. We analyzed differentially expressed genes (DEGs) between sh-Ctrl and sh-TM6SF2 cells by using the “edgeR” package based on the R platform[21]. Fold change > 1.2 and adjustedP-value < 0.05 were set as the screening cutoffs for the upregulated DEGs. These DEGs were further analyzedviaDAVID 6.8 (https://david.ncifcrf.gov/)for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. The “ggplot2” package in R is used for result visualization.
Preparation of fatty acid solution
A 5 mmol/L stock solution of PA (Sigma-Aldrich) was obtained by dissolving PA in 3% of bovine serum albumin (BSA, Sigma-Aldrich) medium by continuous stirring for approximately 4 h in a 60 °C water bath. The stock solution was then diluted in DMEM to achieve the designated concentration, and 3% of BSA was used as a control. The concentrations of free fatty acids range from 0.1 to 0.7 mmol/L[22-24].
Glucose and insulin tolerance test
Glucose tolerance test (GTT) and insulin tolerance test (ITT) were performed as previously described[25]. After the above steps were completed, the glucose concentrations of the blood collected from the tail vein at the indicated points were analyzed immediately using a glucose meter (Yuwell, China).
Nile red staining
After sh-ctrl and sh-TM6SF2 L02 cells were constructed, they were incubated in DMEM containing 150 μmol/L of PA for 24 h to generate a steatosis cell model. Cells were then fixed with 4% paraformaldehyde and incubated with 0.1 μg/mL Nile Red for 15 min. After washing with PBS, the cells were incubated with DAPI (Solarbio) for 5 min. Images were taken with a Leica microscope (Leica, Germany)and quantified using the ImageJ software.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) imaging was performed using a 7.0 T Bruker BioSpec MRI scanner(Bruker, United States). Before imaging, the mice were fasted overnight to avoid disturbance of stomach contents during liver imaging. The mice were first anesthetized with oxygen containing 2%–3%isoflurane and then placed in a prone position with their heads facing inward. Throughout the imaging process, the mice were continuously provided with oxygen mixed with 1%-3% isoflurane through the nosecone to maintain the respiratory rate at 50-70 breaths/minute. All mice underwent abdominal MRI to determine the maximum cross-sectional area of the liver. The region of interest was analyzed using ImageJ software, and the size of the cross-sectional area was represented by the number of pixels.
Histological analysis
Liver sections were embedded in paraffin and stained with hematoxylin and eosin (H&E), and frozen liver sections were stained with Oil Red O (O8010-5g; Solarbio) for lipid visualization. Images were obtained using a light microscope (Leica). Oil Red O- stained sections were quantified using ImageJ software.
FAO measurement
A total of 6000 cells were seeded in an XF96 cell culture microplate, and each group was assayed in five or six replicate wells. After cell adherence (about 8 h), the growth medium was changed to a substratelimited growth medium (0.5 mmol/L of glucose, 1 mmol/L of GlutaMAX, 0.5 mmol/L of XF Lcarnitine, 1% FBS, pH 7.4) for overnight incubation. Before the Seahorse Bioscience XF96 instrument(Agilent, United States) was started, the medium was changed to a PA measurement medium. Different inhibitors (oligomycin 2 μmol/L, FCCP 2 μmol/L, and R/A 1.2 μmol/L) were added to each chamber when the instrument began to measure the oxygen change rate. The difference in each group’s oxygen consumption rate (OCR) revealed the level of fatty acid oxidation (FAO) based on palmitate substrates.The basal respiration, maximal respiration, spare respiratory capacity, and ATP production were calculated.
Cell viability assay
Cell viability was assessed using the CCK-8 assay (NCM Biotech, China). Sh-ctrl and sh-TM6SF2 cells were seeded in a 96-well plate at a density of 5000 cellsperwell, and a cell viability assay was performed after PA or fatty acid-free BSA treatment for 24 h. Absorbance (450 nm) was measured using a microplate reader (BioTek, United States). Each sample was assayed in five replicate wells, and the experiment was independently performed three times.
Quantitation of intracellular TG levels in cell lines
After PA or BSA treatment for 24 h, cells were lysed on ice with RIPA buffer (Beyotime, Beijing, China)for 20 min and centrifuged at 13000 rpm for 20 min at 4 °C. Then the supernatant was transferred to a new tube. TG levels were evaluated using a TG detection kit (Nanjing Jiancheng, Jiangsu, China) and normalized to the total protein level. The experiment was independently performed three times.
Bioinformatic analyses
Expression profiles of TM6SF2 in NAFLD cases were retrieved from the Gene Expression Omnibus(GEO; www.ncbi.nlm.nih.gov/geo) database. The accession numbers are GSE130970, GSE48452,GSE83452, and GSE89632. The details of the cohorts are presented in Supplementary Table 2.
Statistical analysis
All data are presented as the mean ± SD. Student’s t-test was used to evaluate the differences between the two groups. Differences between more than two groups were compared using one-way analysis of variance followed by Tukey’s multiple comparisons test.In vitrostudies, all experiments were independently performed three times in vitro. The SPSS software (version 25.0) was used for all statistical analyses.P< 0.05 was considered statistically significant and denoted asaP< 0.05,bP< 0.01,andcP< 0.001; NS: Not significant.
RESULTS
Hepatic TM6SF2 expression is upregulated in patients with NAFLD and HFD-induced mice models
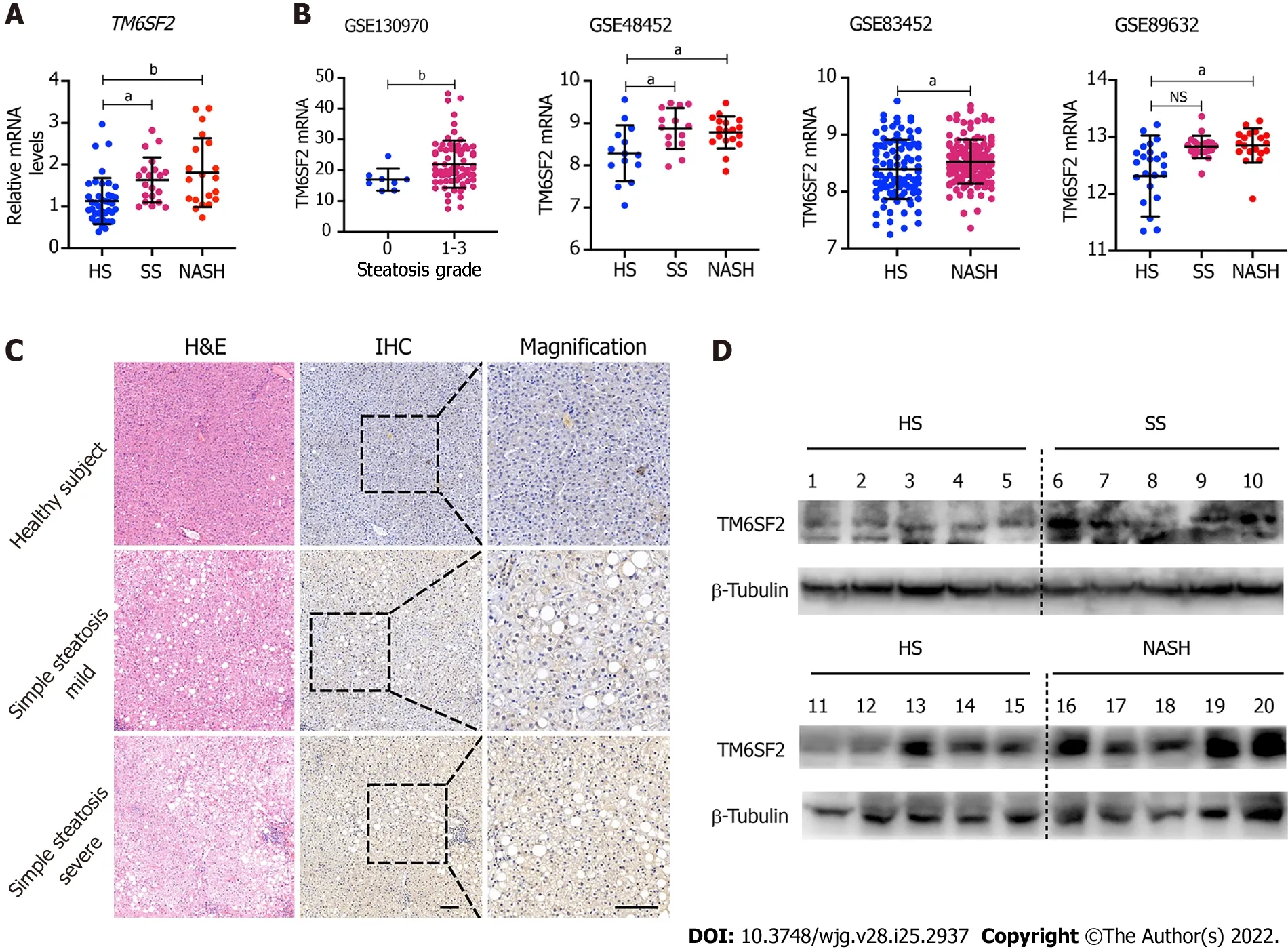
Figure 1 Hepatic TM6SF2 expression is upregulated in nonalcoholic fatty liver disease patients. A: Hepatic mRNA levels of TM6SF2 in liver specimens, including healthy subjects (HS, n = 40) and patients with simple steatosis (SS, n = 20) or nonalcoholic steatohepatitis (NASH, n = 20); B: Hepatic mRNA levels of TM6SF2 in individuals with or without nonalcoholic fatty liver disease (NAFLD) from four expression microarray series retracted from Gene Expression Omnibus database; C: Immunohistochemical staining with TM6SF2 was performed on individuals with or without NAFLD. Representative images of normal liver tissues vs tissues of SS were shown. Scale bars: 50 μm; D: Immunoblot analysis of TM6SF2 expression in liver specimens. HS (n = 10), SS (n = 5) and NASH (n =5). aP < 0.05; bP < 0.01, NS: Not significant; HS: Healthy subjects; SS: Simple steatosis; NASH: Nonalcoholic steatohepatitis; IHC: Immunohistochemistry; H&E:Hematoxylin and eosin.
The mRNA levels of TM6SF2 were examined in 80 liver samples of patients with NAFLD and healthy subjects (n= 40), which showed that the transcriptional levels of TM6SF2 were both elevated in patients with SS or nonalcoholic steatohepatitis (NASH) (Figure 1A, SS:n= 20; NASH:n= 20). Additionally, we retrospectively analyzed another four transcriptomic datasets retrieved from the GEO database(GSE13970, GSE48452, GSE83452, and GSE89632, Figure 1B), which contained transcriptional profiles of both healthy participants and patients with NAFLD. Patients with SS or NASH displayed higher hepatic TM6SF2 mRNA levels than healthy participants. Immunohistochemistry staining indicated that TM6SF2 tended to upregulate TM6SF2 expression from normal liver to severe SS (Figure 1C). We also noted upregulation of TM6SF2 protein in the liver samples of patients with SS or NASH (Figure 1D). To further confirm TM6SF2 overexpression under overnutrition, we evaluated the changes in TM6SF2 expression in andin vitroNAFLD cell model and stimulated two cell lines, L02 and HepG2, with PA, as it was reported that thisin vitromodel mimics steatosisin vivo[22-24]. The results showed that PA stimulation increased the protein levels of TM6SF2 in a time- and dose-dependent manner(Supplementary Figure 1A). Further, whether in protein or mRNA levels, the augmented TM6SF2 expression was also demonstrated in HFD-fed mice for 12 wk (Supplementary Figure 1B). These results revealed that hepatic TM6SF2 levels were upregulated in patients with NAFLD and HFD-induced mouse models.
TM6SF2 overexpression mitigates hepatic lipid accumulation in HFD-induced mice models
As we had found that the levels of TM6SF2 were increased in participants with NAFLD and HFDinduced models, we wanted to determine the physiological significance of TM6SF2 elevation when NAFLD phenotypes occur. To this end, we used the AAV8 vector to deliver human TM6SF2 (AAV8-TM6SF2) in the livers of mice (AAV8-vector was used as a control) and fed them HFD for 16 wk(Figure 2A and B). The results showed that TM6SF2 overexpression in the liver significantly reduced the liver/body weight ratio, and its impact on body weight was limited (Figure 2C and D). Hepatic TM6SF2 overexpression in mice resulted in lower levels of hepatic lipid content as well as lower serum ALT and AST levels (Figure 2E and F). TM6SF2-overexpression groups relative to controls displayed equal body size (Figure 2G) but demonstrated a minor fatty liver and a smaller cross-sectional area of the fatty liver(Figure 2H). Hepatic lipid accumulation was also ameliorated in AAV8-TM6SF2 mice, as indicated by the pathological examination (Figure 2I). We also observed that TM6SF2 knocked-down cells reconstituted with wild-type TM6SF2 exhibited decreased intracellular lipid accumulation and improved PA-induced cell death (Supplementary Figure 2). This evidence suggests that the elevation of TM6SF2 is a potential reaction to antagonize NAFLD phenotypes and serves a protective role during NAFLD.
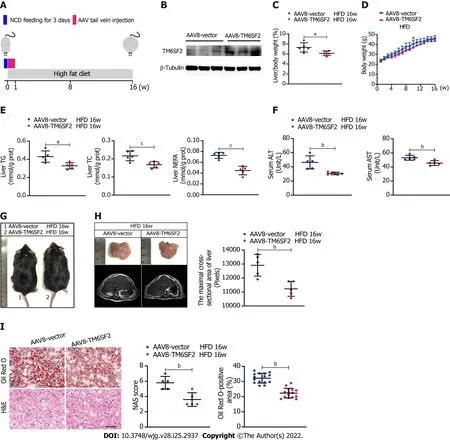
Figure 2 Overexpression of TM6SF2 improves hepatic lipid accumulation in high-fat diet-induced mice models. A: Schematic representation of animal experiments. Mice were injected with AAV8-vector or AAV8-TM6SF2 virus via the tail vein and fed a high-fat diet for 16 wk (n = 5 mice per group); B: The efficiency of TM6SF2 overexpression in liver were shown; C-F: The liver/body weight ratio (C), the body weight (D), hepatic lipid content (E), and serum levels of alanine aminotransferase and aspartate transaminase (F) were shown; G: Representative images of gross morphology; H: Left, representative photos of fatty livers(top) and livers subjected to magnetic resonance imaging scanning (bottom) were shown. Right, the maximal cross-sectional area of livers is quantified by the number of pixels; I: Left, representative images of hematoxylin and eosin-staining (left) and Oil Red O-staining (right) of liver sections. Right, the nonalcoholic fatty liver disease activity score (left) and quantification of lipid (right, 8 fields of each mouse were examined) were performed. aP < 0.05; bP < 0.01; cP < 0.001. H&E:Hematoxylin and eosin; NCD: Normal chow diet; HFD: High-fat diet; NAS: Nonalcoholic fatty liver disease activity score.
Knockdown of TM6SF2 promotes hepatic lipid accumulation and inflammation in the HFD-induced mice model
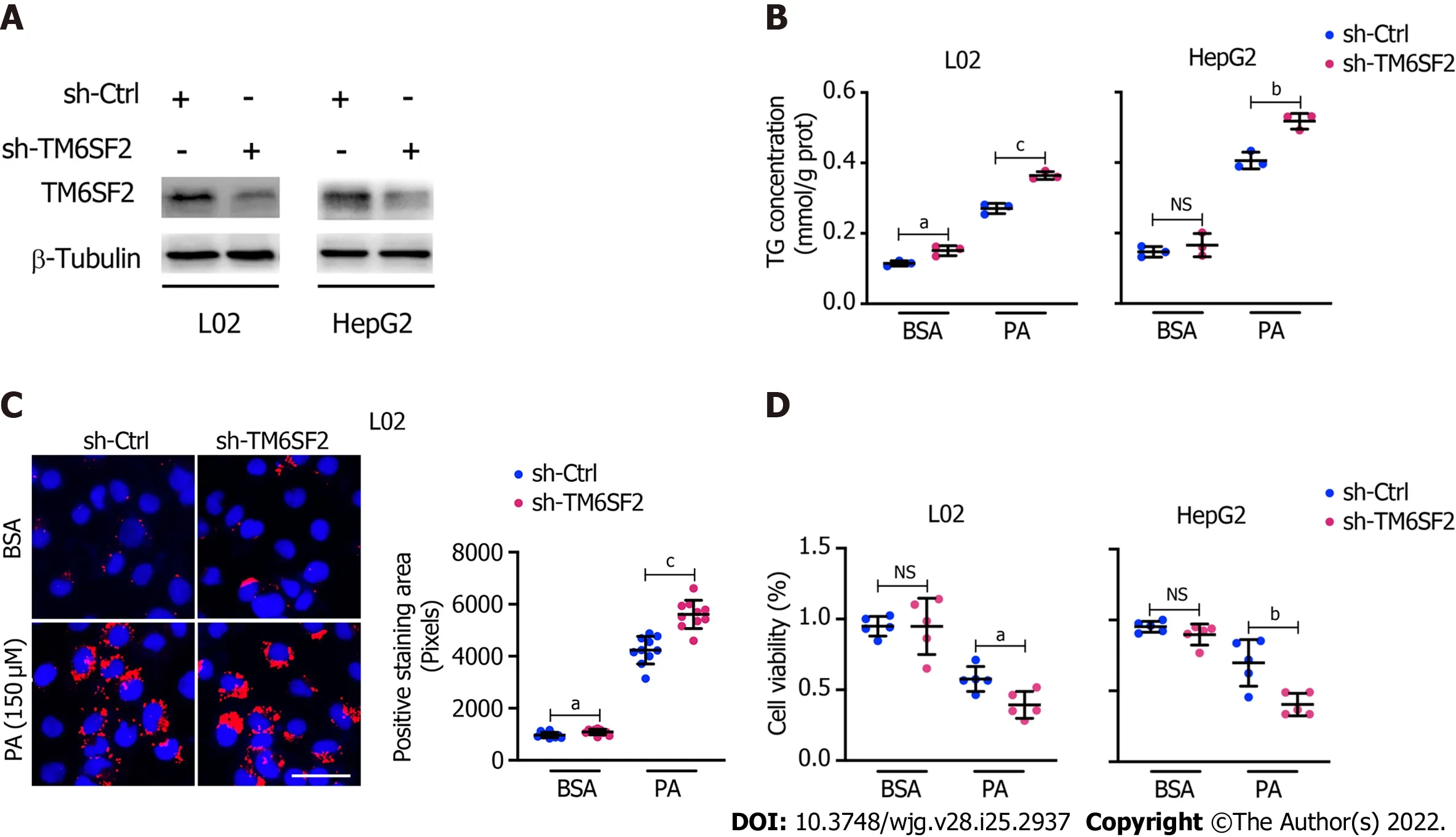
Figure 3 Knockdown of TM6SF2 promotes intracellular lipid accumulation and lipid-overload induced cell death. A: The efficiency of TM6SF2 knockdown in two cell lines; B: Intracellular triglyceride levels (n = 3) in TM6SF2-knockdown cells were examined after palmitic acid (PA) (150 μmol/L) or bovine serum albumin (BSA) (fatty acid free) treatment for 24 h; C: Nile red staining (left, nuclei labeled with DAPI, blue) and quantification of lipid accumulation (right) of sh-Ctrl or sh-TM6SF2 L02 cells after PA or BSA treatment. Scale bars: 50 μm; D: TM6SF2-knockdown cells were incubated with PA (150 μmol/L) or BSA for 24 h. The cellular viability was examined (n = 5). aP < 0.05; bP < 0.01; cP < 0.001. NS: Not significant; BSA: Bovine serum albumin; PA: Palmitic acid; TG: Triglyceride; μM:μmol/L.
Pathologically, there exists a genetic E167K mutation in the TM6SF2 protein in humans, and those with the TM6SF2 E167K mutation are associated with an increased risk of NAFLD[26]. Ehrhardtet al[27]reported that the E167K mutation causes downregulation of TM6SF2 at the protein level. To simulate the E167K condition and explore the influence of TM6SF2 on lipid accumulation, we generated TM6SF2-knockdown cells in both the L02 and HepG2 cell lines (Figure 3A), which were incubated with PA (150 μmol/L) or BSA (fatty acid free) for 24 h. We found that TM6SF2-knockdown cells demonstrated higher levels of intracellular TG (Figure 3B) and more severe lipid accumulation (Figure 3C). Lipid overload induces endoplasmic reticulum stress (ERS) and causes cell death[28,29]. The cell viability assay results proved that TM6SF2 knockdown markedly exacerbated PA-induced cell death (Figure 3D), which reflected the increase in lipid content in the TM6SF2-knockdown group. To determine whether TM6SF2 deficiency exacerbated hepatic steatosisin vivo, we generated hepatic TM6SF2-knockdown (hereafter referred to as AAV-shTm6sf2, AAV-shNC was used as a control) mice by injecting AAV vectors carrying TM6SF2-targeting shRNA, and mice were fed an HFD or NCD for 16 wk (Figure 4A and B).Compared to the control mice, the HFD-fed AAV-shTm6sf2 mice displayed severe hepatocellular ballooning degeneration and lipid accumulation, as shown by H&E and Oil Red O staining (Figure 4C);these mice had higher levels of hepatic TG, TC, and NEFA (Figure 4D). Similarly, AAV-shTm6sf2 mice had a higher liver/body weight ratio (Figure 4E) but exhibited a slightly higher body weight (Figure 4F)only at the initiation of feeding. In addition, hepatic TM6SF2-knockdown mice relative to controls displayed a modest increase in size (Figure 4G) but a significantly enlarged liver and a larger maximal cross-sectional area of the liver (Figure 4H). However, these parameters did not differ between the two groups when placed in a NCD environment (Figures 4D–F).
It is noteworthy that hepatic steatosis is often accompanied by inflammation, and the boundary between the two phases (SS and NASH) of NAFLD is blurred[30]. The hepatic TM6SF2-knockdown group displayed higher serum levels of inflammatory cytokines, such as TNF, IL-6, IL-1β, CCL2, and CXCL10 than control mice after 16 wk of HFD feeding (Supplementary Figure 3A). Consistently, hepatic expression of inflammation-related genes was also augmented in the AAV-shTm6sf2 group(Supplementary Figure 3B). Meanwhile, the AAV-shTm6sf2 group showed higher serum levels of ALT and AST (Supplementary Figure 3C) and more infiltrated neutrophils and macrophages than the AAVshNC group (Supplementary Figure 3D). These results show that TM6SF2 knockdown promotes hepatic lipid accumulation and inflammation in an HFD-induced mouse model.
Hepatic knockdown of TM6SF2 enhances de novo lipogenesis
Triplicate samples of PA-treated TM6SF2-knockdown L02 cells and their controls were used for transcriptome analysis to explore the changes in biological processes in TM6SF2 knockdown and identify upregulated DEGs. KEGG pathway enrichment analysis was applied to explore the function of these DEGs, which highlighted that fatty acid metabolism was the key process that changed in TM6SF2-knockdown cells (Figure 5A), and genes associated with fatty acid synthesis were found to be upregulated (Figure 5B). To confirm this,in vitroNAFLD models were used to verify our findings. PA is a potent agonist of fatty acid synthesis[31]. PA administration activated fatty acid synthesisin vitro, as indicated by increased protein levels of p-ACC, FASN, SCD-1, and sterol regulatory element-binding protein 1c (SREBP-1c) (Figure 5C). When comparing the levels of these proteins between TM6SF2-knockdown cells and the corresponding controls, the results showed that these proteins were upregulated in TM6SF2-knockdown cells, suggesting that PA-induced fatty acid synthesis was enhanced by the knockdown of TM6SF2 (Figure 5D).De novolipogenesis is mainly mediated by SREBP-1c on the transcriptional regulation of ACC and fatty acid synthase. The mature form of SREBP-1c translocates to the nucleus and activates its target genes, such asAcacaandFasn[32]. This translocationto-nucleus phenomenon was also enhanced in the TM6SF2-knockdown cells (Figure 5E). In addition,TM6SF2 knockdown increased hepatic malonyl-CoA, a metabolite of ACC[33], in HFD-fed AAVshTm6sf2 mice (Figure 5F), which indicated that ACC activity was enhanced due to TM6SF2 deficiency.It is noteworthy that the transcriptional activity of hepatic SREBP-1c is also influenced by circulating insulin status. We evaluated the mice’s blood glucose and insulin levels and found no differences between the two groups with or without HFD feeding (Supplementary Figure 4A). In addition, no obvious differences were noted in the GTT and ITT (Supplementary Figure 4B). These results suggest that the enhanced steatosis in hepatic TM6SF2-knockdown mice is unlikely to be induced by altering insulin sensitivity. Moreover, we also noted that TM6SF2 knockdown did not affect the fatty acid transport process but caused an increase in the mRNA levels of molecules involved in fatty acid uptake(e.g., CD36 and FABP1) (Figure 5B). This increase was augmented when the TM6SF2-knockdown cells were under starvation or hypoxic conditions (Supplementary Figure 4C), as hypoxia and starvation have been reported to enhance lipid uptake[34-36]. In parallel, the levels of fatty acid synthesis- and uptake-related genes were elevated in the livers of AAV-shTm6sf2 mice compared to control mice(Figure 5G). This evidence suggests that the unfavorable fatty liver in hepatic TM6SF2-knowndown mice may be associated with enhancedde novolipogenesis and lipid uptake.
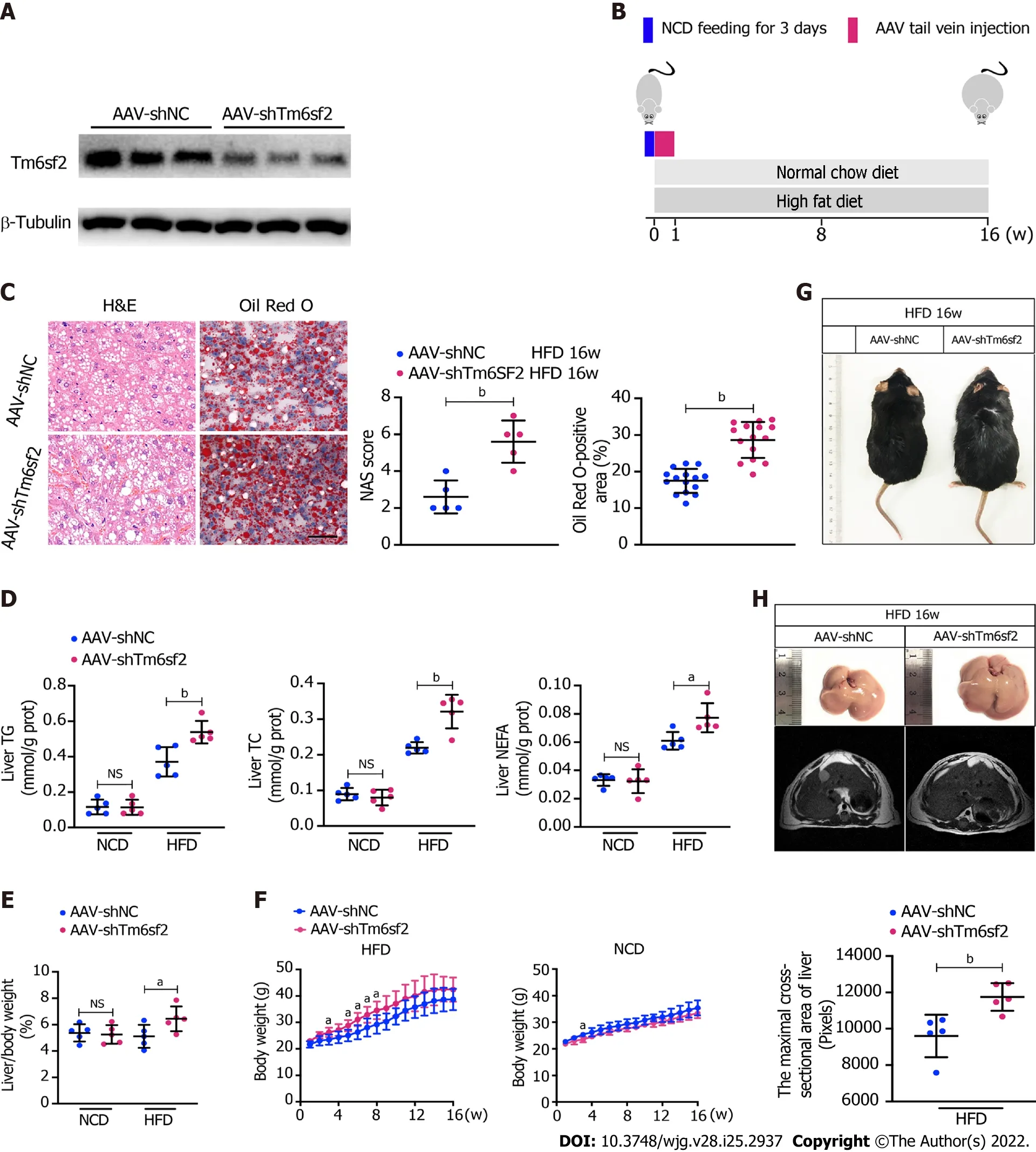
Figure 4 TM6SF2 deficiency exacerbates hepatic lipid accumulation and inflammation. A: Mice were injected with AAV-shNC and AAV-shTm6sf2 viruses via the tail vein and fed a high-fat diet or normal chow diet for 16 wk (n = 5 mice per group). The efficiency of TM6SF2 knockdown in liver were shown; B:Schematic representation of animal experiments; C: Left, representative images of hematoxylin and eosin-staining (left) and Oil Red O-staining (right) of liver sections. Right, the Nonalcoholic fatty liver disease activity score (left) and quantification of lipid (right, 8 fields of each mouse were examined) were performed; D-F:Hepatic lipid contents (triglyceride, total cholesterol and non-esterified fatty acids) (D), liver/body weight ratio (E) and body weight (F) were measured; G:Representative images of mice morphology; H: Top, representative photos of fatty livers (top) and livers subjected to magnetic resonance imaging scanning (bottom)of mice were shown. Bottom, the maximal cross-sectional area of livers is quantified by the number of pixels. aP < 0.05; bP < 0.01. NS: Not significant; H&E:Hematoxylin and eosin; NCD: Normal chow diet; HFD: High-fat diet; TG: Triglyceride; TC: Total cholesterol; NEFA: Non-esterified fatty acids.
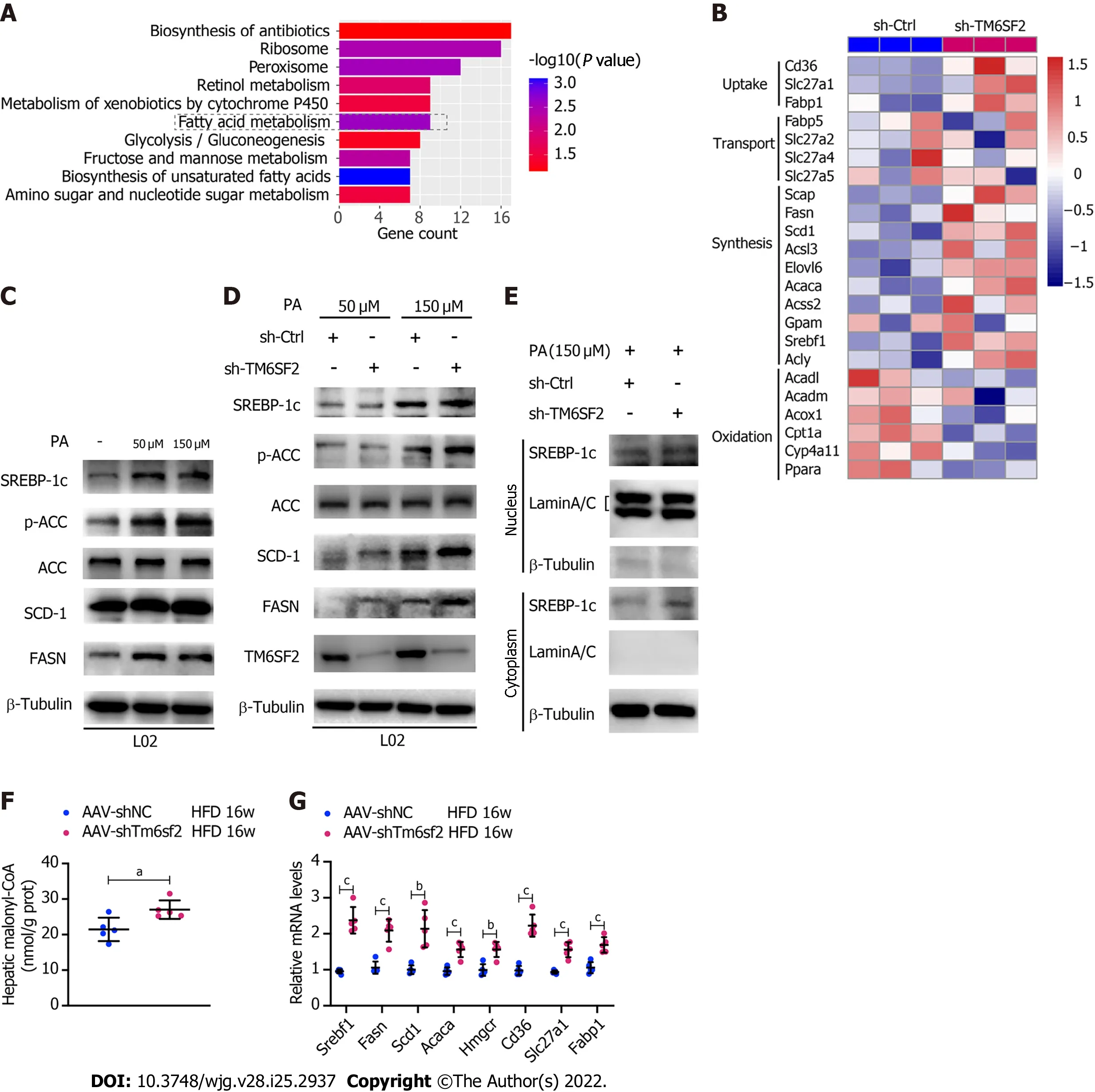
Figure 5 The activity of fatty acid synthesis was enhanced in TM6SF2 knockdown group. A: Pathway enrichment of the up-regulated differentially expressed genes by Kyoto Encyclopedia of Genes and Genomes analysis; B: Microarray heatmap of genes involved in fatty acid metabolism in TM6SF2-knockdown cells; C: The SREBP-1c target protein levels in L02 and HepG2 cells after palmitic acid (PA) treatment; D: The SREBP-1c target protein levels in TM6SF2-knockdown cells after PA treatment; E: Immunoblot analysis of SREBP-1c in cytoplasmic and nuclear extracts; F: Hepatic malonyl-CoA contents of mice in two groups (n = 5 mice per group); G: The mRNA levels of genes involved in fatty acid synthesis (Srebf1, Fasn, Scd1, Acaca and Hmgcr) and uptake (Cd36, Slc27a1 and Fabp1). aP <0.05; bP < 0.01; cP < 0.001. HFD: High-fat diet; PA: Palmitic acid; ACC: Acetyl-CoA carboxylase; μM: μmol/L.
TM6SF2 deficiency could lead to impaired FAO
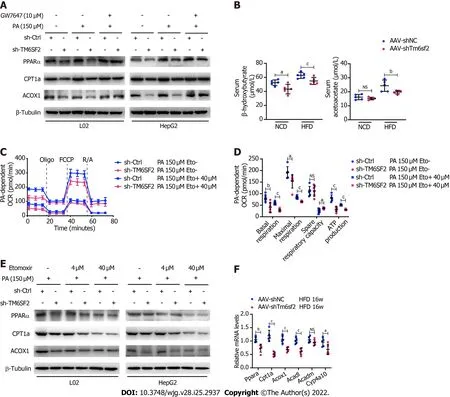
Figure 6 TM6SF2 deficiency caused the downregulation of fatty acid oxidation in liver. A: Protein levels of PPARα, CPT1A and ACOX1 was analyzed in TM6SF2-knockdown cells after palmitic acid (PA) stimulation and their response to GW7647 (10 μmol/L) treatment; B: Serum levels of β-hydroxybutyrate and acetoacetate in normal chow diet- or high-fat diet-fed AAV-shNC or AAV-shTm6sf2 mice (n = 5 mice per group); C and D: PA-dependent oxygen consumption rate in sh-Ctrl and sh-TM6SF2 L02 cells with or without etomoxir (ETO, 40 μmol/L) administration (C, n = 6). The basal respiration, maximal respiration, spare respiratory capacity, and ATP production were calculated (D); E: Protein levels of PPARα, CPT1A and ACOX1 were determined in TM6SF2-knockdown cells with or without ETO treatment under PA (150 μmol/L) stimulation; F: The hepatic mRNA levels of fatty acid oxidation-related genes in the indicated mice. aP < 0.05; bP <0.01; cP < 0.001. PA: Palmitic acid; NS: Not significant; NCD: Normal chow diet; HFD: High-fat diet; OCR: Oxygen consumption rate; μM: μmol/L.
It was previously reported that NAFLD caused by TM6SF2 deficiency was partially due to impaired VLDL secretion, which is detrimental to the liver, as VLDL secretion is considered a significant pathway of lipid export in the liver[18]. FAO, another method of energy expenditure[37], has rarely been reported in the absence of the TM6SF2 protein. Therefore, we evaluated and compared the expression of the key molecules involved in FAO. The results showed that TM6SF2-knockdown cells exhibited decreased levels of FAO-related proteins[38], including PPARα, CPT1A, and ACOX1, with or without PA stimulation (Figure 6A), which is consistent with the results of transcriptomic data (Figure 5B).Furthermore, PPARα signaling has been reported to function as a key factor in regulating FAO[39], and we found that the downregulated levels of FAO-related proteins in TM6SF2-knockdown cells could be reversed by PPARα activation through PPARα agonist (GW7647) stimulation (Figure 6A). We next tested hepatic FAOin vivoby evaluating the levels of serum ketone bodies, β-OH, and acetoacetate in both AAV-shTm6sf2 mice and control groups after a 16 h fast. The results showed that the levels of ketone bodies were significantly lower in the TM6SF2-knockdown group (Figure 6B), suggesting that TM6SF2 deficiency reduces FAO. In addition, compared to the control group, TM6SF2-knockdown L02 cells had a lower OCR with PA as the only substrate (Figures 6C and D). Meanwhile, the CPT1A inhibitor etomoxir inhibited the FAO process and diminished the difference in both groups, as well as the FAO-related proteins (Figure 6C-E). AAV-shTm6sf2 mice also showed a lower expression of FAOrelated genes in the liver (Figure 6F). Together, these data indicate that TM6SF2 deficiency impairs cellular FAO.
MK4074 is effective in treating NAFLD exacerbated by TM6SF2 deficiency
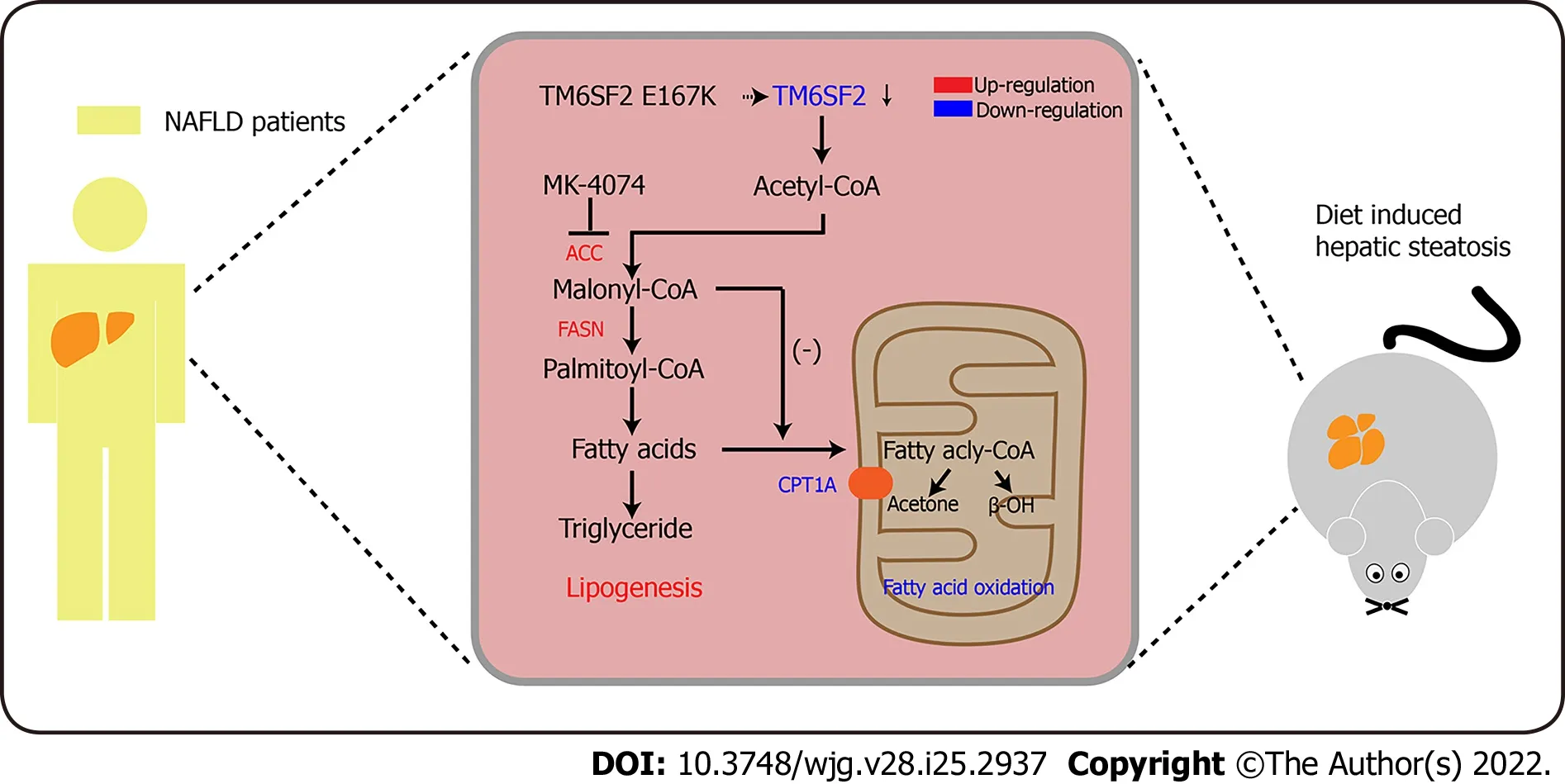
Figure 7 Schematic diagram of MK-4074 blocking acetyl-CoA carboxylase pathway and alleviating nonalcoholic fatty liver disease in TM6SF2-deficient mice. ACC: Acetyl-CoA carboxylase; NAFLD: Nonalcoholic fatty liver disease.
These results showed that TM6SF2 deficiency could lead to abnormal fatty acid metabolism within cells,characterized by enhanced lipogenesis and uptake, accompanied by impaired fatty acid β-oxidation(Figure 7). It is worth noting that the suppressed β-oxidation process may be associated with the increased malonyl-CoA content due to the enhanced activity of ACC in this situation. Malonyl-CoA is a natural CPT1A inhibitor, which causes a decrease in β-oxidation in mitochondria[40]. Therefore,inhibiting the activity of ACC may be a promising method to alleviate fatty liver disease in mice with hepatic TM6SF2 deficiency. To test this idea and rectify this metabolic abnormality, we thought of a way of inhibiting ACC, this rate-limiting enzyme, by using a previously reported ACC inhibitor, MK-4074,which could simultaneously limit thede novolipid synthesis pathway and increase the level of intracellular lipid oxidation, to explore whether the drug has therapeutic effect on TM6SF2-deficient cases. To this end, both AAV-shTm6sf2 and control mice were first subjected to 8-wk HFD feeding to induce NAFLD phenotypes, and then two groups of mice received MK-4074 (MK-4074, 10 mg/kg/day)or placebo (normal saline) treatment and were fed with HFD for an additional eight weeks (Figure 8A).The results showed that MK-4074 treatment could diminish the increase in the levels of malonyl-CoA(Figure 8B), as well as hepatic TG, TC, and NEFA caused by TM6SF2 deficiency (Figure 8C), suggesting the success of blocking the SPREBP-1c/ACC pathway. Compared to placebo treatment, the HFD-fed AAV sh-Tm6sf2- mice under MK-4074 treatment exhibited mild hepatic ballooning degeneration and steatosis (Figure 8D). At the same time, MK-4074 treatment improved the liver/body weight ratio(Figure 8E), although the body weight improvement was rather limited (Figure 8F). In addition, the beneficial effect is also manifestedin vitroexperiments that MK-4074 could reduce lipid accumulation(Figure 9A and B) and improve PA-induced cell death in TM6SF2 knocked-down groups (Figure 9C).Meanwhile, MK-4074 inhibited malonyl-CoA production (Figure 8B) and improved the decrease in FAO caused by TM6SF2 deficiency, as evidenced by the increased FAO-related protein and ketone body levels in the TM6SF2-knockdown group (Figure 9D and E), which renders MK-4074 the prime option for treating this condition.
DISCUSSION
The present study revealed that hepatic TM6SF2 expression was upregulated in both NAFLD mouse models and patients. TM6SF2 overexpression alleviated NAFLD phenotypes in HFD-induced models,suggesting a protective role for reactive TM6SF2 elevation under NAFLD conditions. Simulating the situation in patients with the E167K mutation, TM6SF2 knockdown exacerbated hepatic steatosis and inflammation. We noted a dysregulated fatty acid metabolism process under TM6SF2 deficiency,characterized by enhanced fatty acid uptake and fatty acid synthesis, accompanied by an impaired FAO process and insufficient ketone body production. Given the potential impact of TM6SF2 on fatty acid metabolism in NAFLD, exploring the effects of targeted NAFLD therapeutics on fatty liver is of the utmost importance, inspiring us to consider the impact of genetic variation on fatty liver. We used a previously reported liver-specific ACC inhibitor, MK-4074, which could reduce hepatic lipogenesis while increasing FAO, to explore the therapeutic effect on fatty liver caused by TM6SF2 deficiency(Figure 7). Our results show that MK-4074 has an encouraging effect on treating TM6SF2 knockdowninduced NAFLD, as indicated by the reduced hepatic lipid content, lower liver/body ratio, and alleviated PA-induced cell death suggesting the effectiveness of MK-4074 in treating this condition.
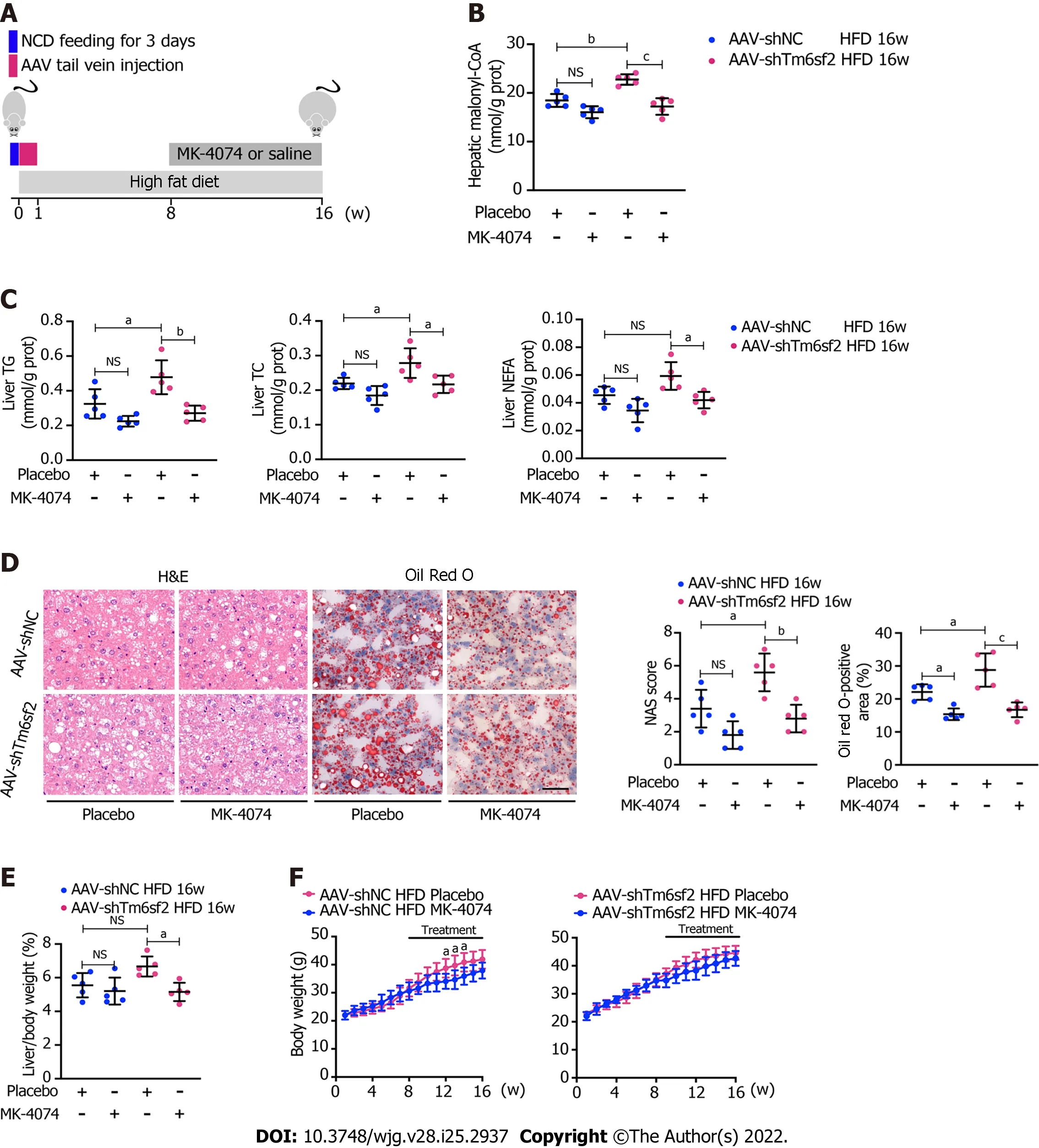
Figure 8 Therapeutic potential of MK-4074 on nonalcoholic fatty liver disease caused by TM6SF2 deficiency. A: Schematic representation of animal experiments; B-D: Two groups (AAV-shNC and AAV-shTm6sf2) of mice were fed a high-fat diet (HFD) for 8 wk to induce nonalcoholic fatty liver disease phenotypes and then each subgroup was dosed with MK-4074 (10 mg/kg/day) or placebo orally for additional 8 wk. The hepatic malonyl-CoA levels (B) and lipid content (C) as well as results of hematoxylin and eosin-staining and Oil Red O-staining of liver sections (D) were shown; E and F: The liver/body weight ratio (E) and the body weight (F) of HFD-fed AAV-shNC and AAV-shTm6sf2 mice with or without MK-4074 treatment (n = 5 mice per group) were shown. aP < 0.05; bP < 0.01; cP <0.001. NS: Not significant; H&E: Hematoxylin and eosin; NCD: Normal chow diet; HFD: High-fat diet; TG: Triglyceride; TC: Total cholesterol; NEFA: Non-esterified fatty acids.
Previous studies have considered TM6SF2 as a pivotal molecule in hepatic lipid output in the form of VLDL-TG, and a majority of studies have focused on the association between the export of accumulated lipids and the TM6SF2 E167K mutant[15,18,41]. The TM6SF2 E167K mutant may result in a misfolded protein easily degraded within the cells, resulting in decreased TM6SF2 levels[42]. The liver is a pivotal hub for lipid metabolism, and its dysregulation could be a significant cause of NAFLD[43]. Therefore,we investigated the cause of TM6SF2 deficiency-induced NAFLD from another perspective, dysregulated fatty acid metabolism. Our results revealed that TM6SF2 knockdown significantly increased the expression of SREBP-1c and its target genes bothin vivoandin vitro, suggesting abnormally enhancedde novolipogenesis (DNL). Meanwhile, the results showed that the levels of OCR were decreased in the TM6SF2-knockdown groups, indicating that the FAO process was impaired under these circumstances.We further tested the transcriptional levels of FAO-related genes in hepatic TM6SF2-knockdown mice fed an HFD and showed lower levels of genes involving FAO than the control mice on the same diet for 16 wk. Previous studies showed that elevated levels of malonyl-CoA would suppress FAO by inhibiting CPT1A activity[44]. In our study, the malonyl-CoA content was higher in AAV-shTm6sf2 mice than in the control mice, which implies that the decreased FAO process is related to the enhanced DNL due to TM6SF2 knockdown.
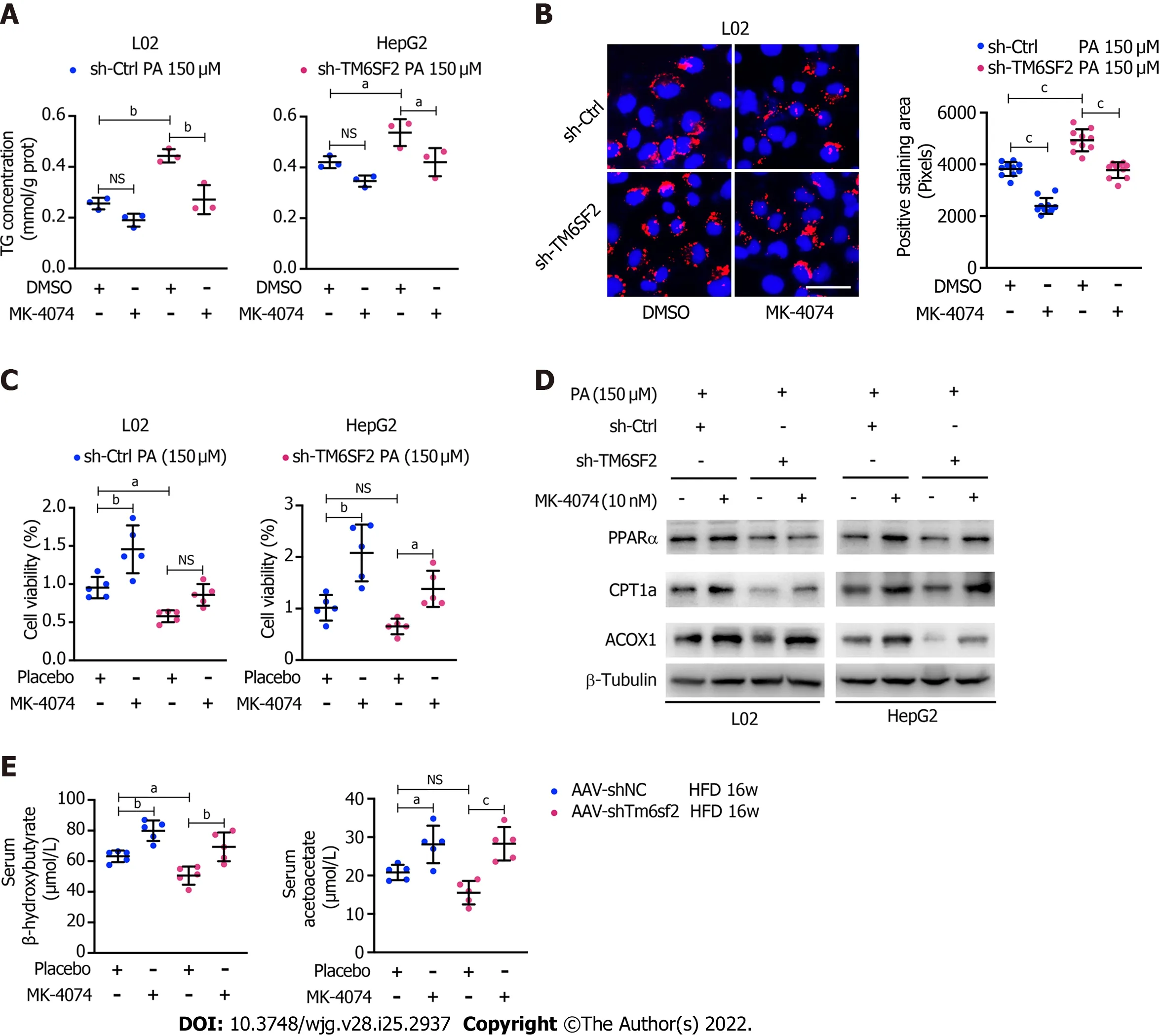
Figure 9 MK-4074 can improve intracellular lipid accumulation, lipid overload-induced cell death, and fatty acids β-oxidation. A: Intracellular triglyceride levels (n = 3) in TM6SF2-knockdown cells with palmitic acid (PA) (150 μmol/L) stimulation were examined after MK-4074 (10 nM) treatment for 24 h; B:Representative nile red staining images (left) and lipid droplet quantification (right) of sh-Ctrl or sh-TM6SF2 L02 cells after PA stimulation in response to MK-4074 (10 nM) treatment (10 fields of each sample were examined). Scale bars: 50 μm; C: The cellular viability was examined in TM6SF2-knockdown cells with or without MK-4074 treatment in response to PA-induced cell death (n = 5); D: The sh-Ctrl and sh-TM6SF2 cells were cultured with PA (150 μmol/L) for 24 h. Cells were then treated with MK-4074 (10 nM) for 12 h. The expression of fatty acid oxidation-related proteins was determined; E: Serum levels of β-hydroxybutyrate and acetoacetate in the indicated mice with or without MK-4074 treatment. aP < 0.05; bP < 0.01; cP < 0.001. NS: Not significant; HFD: High-fat diet; TG: Triglyceride; PA: Palmitic acid; μM:μmol/L.
Currently, genetic technologies have already allowed for the identification of a subgroup of people with an increased risk of NAFLD or NAFLD-related complications associated with specific pathophysiological characteristics[45]. Unfortunately, there is no precise treatment for metabolic diseases with specific congenital genetic variations in NAFLD. However, much effort has been made to elucidate the mechanism of NAFLD caused by genetic mutations and implement precision medicine. For example,human genetics of NAFLD offers the opportunity to apply targeted therapy with the PNPLA3 I148M variant. This locus is the strongest genetic factor that increases susceptibility to NAFLD[10]. Recently,researchers used mice with the PNPLA3 variant knock-in and tried to improve NAFLD phenotypes through antisense oligonucleotides to downregulate the PNPLA3 mutant protein[46]. Although the mechanisms are not fully understood, accumulating evidence reveals that PNPLA3 I148M impairs lipid droplet remodeling in hepatocytes due to the loss of lipase activity[47]. Inhibition of PNPLA3148M expression reduced liver fat content in these engineered mice and, specifically, had a beneficial effect in mitigating inflammation and fibrosis. If these applications are translated into humans, after safety and reliability examinations, downregulation of PNPLA3148M may be the first case of human geneticsbased targeted therapy. The importance of this method is to treat not only the phenotype of the disease but also the cause.
As described in the PNPLA3 mutant protein case, this evidence proved the possibility of targeted therapy to eliminate the cause of fatty liver in patients carrying a specific risk allele. However, there have been no reports of attempts to treat fatty liver caused by TM6SF2 deficiency or its E167K mutant.Based on our data, mice with TM6SF2 deficiency show enhanced DNL. Among the TM6SF2/SREBP-1c/ACC axis, ACC acts as the rate-limiting enzyme for converting acetyl-CoA to malonyl-CoA[48]. The product, malonyl-CoA, was further catalyzed by FASN to form palmitate. TGs are one of the many lipid species that are integrated with DNL-derived palmitate. ACC inhibitors have the potential to mitigate hepatic steatosis in NAFLD, given that ACC is essential and pivotal in hepatic fatty acid metabolism.Independent studies have revealed that blocking DNL by ACC inhibition has a beneficial effect in reducing liver fat, steatosis, and inflammation in NAFLD models[44,49]. Among ACC inhibitors, MK-4074 has recently been tested for NAFLD treatment, and its effects have been encouraging. MK-4074 treatment decreased DNL and enhanced FAO in participants under overnutrition. Based on its function,we hypothesized that it could be used to treat fatty liver caused by the reduction of TM6SF2, andin vivoandin vitroexperiments confirmed this hypothesis. ACC inhibitors have recently been shown to effectively mitigate liver fibrosis in NASH patients[49,50]. The accumulating evidence has shown that TM6SF2 E167K is also associated with NASH and fibrosis[18,51]. Whether ACC inhibitors can be applied to mitigate liver fibrosis in NASH models with TM6SF2 deficiency or the E167K mutant is unknown. This indicates the direction of our future work.
CONCLUSION
This study showed that dysregulated fatty acid metabolism occurs in the context of TM6SF2 deficiency under overnutrition. Therapeutics aimed at abnormal fatty acid metabolism may be a promising strategy for improving the hepatic lipid profile of patients with the TM6SF2 E167K variant in the clinical setting. Furthermore, our study suggests that MK-4074 could be a potential drug to lower hepatic lipid content in a TM6SF2-knockdown NAFLD mouse model. Further investigations are required to test whether this approach would lower hepatic lipid levels in NAFLD patients with the E167K mutation.
ARTICLE HIGHLIGHTS

Research perspectives
TM6SF2 plays a protective role in the HFD condition; its deficiency enhanced hepatic lipid accumulation through dysregulated fatty acid metabolism, and MK-4074 treatment could alleviate the NAFLD phenotypes caused by TM6SF2 deficiency. The next step was to investigate whether MK-4074 has a therapeutic effect in patients with NAFLD harboring the TM6SF2 E167K mutation.
FOOTNOTES
Author contributions:Li ZY and Wu G contributed equally to the work, Li ZY and Wu G performed thein vitroandin vivoexperiments; Qiu C, Zhou ZJ, Wang YP, Song GH and Xiao C performed the analysis of GEO data; Deng GL and Wang RT provided the technical supports; Zhang X interpreted the data; Li ZY wrote the manuscript; Wang XL and Yang YL contributed equally, Wang XL and Yang YL conceived and supervised the study; all authors critically reviewed and edited the manuscript.
Supported byNational Natural Science Foundation of China, No. 81670514 and No. 81702337; Scientific Research Project of Shanghai Municipal Health Commission, No. 202040065; and Natural Science Foundation of Shanghai Scientific and Technological Project of Innovative Action, No. 20ZR1411900.
Institutional review board statement:The study was reviewed and approved by the Shanghai First People's Hospital Institutional Review Board, No. 2016KY074.
Institutional animal care and use committee statement:All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Shanghai First People's Hospital, No. 2020-A0072-01.
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
Data sharing statement:The data used and analyzed during the current study are available from the corresponding author on reasonable request.
ARRIVE guidelines statement:The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORCID number:Zu-Yin Li 0000-0002-3804-0994; Gang Wu 0000-0002-3211-688X; Qiu Chen 0000-0002-5414-1672; Zhi-Jie Zhou 0000-0001-7414-411X; Yu-Peng Wang 0000-0002-9891-2141; Guo-He Song 0000-0002-1452-6316; Chao Xiao 0000-0002-9202-0863; Xin Zhang 0000-0002-8097-4061; Gui-Long Deng 0000-0003-4353-8452; Rui-Tao Wang 0000-0002-3210-8396; Yu-Long Yang 0000-0002-5783-6381; Xiao-Liang Wang 0000-0001-9851-0436.
S-Editor:Fan JR
L-Editor:A
P-Editor:Li X
 World Journal of Gastroenterology2022年25期
World Journal of Gastroenterology2022年25期
- World Journal of Gastroenterology的其它文章
- Non-alcoholic fatty liver disease and the impact of genetic, epigenetic and environmental factors in the offspring
- Role of transcribed ultraconserved regions in gastric cancer and therapeutic perspectives
- Multiple roles for cholinergic signaling in pancreatic diseases
- Early gastric cancer presenting as a typical submucosal tumor cured by endoscopic submucosal dissection:A case report
- Correction to “Aberrant methylation of secreted protein acidic and rich in cysteine gene and its significance in gastric cancer”
- Four-year experience with more than 1000 cases of total laparoscopic liver resection in a single center