
Cloning and Bioinformatics Analysis of TpiA Gene of Vibrio alginolyticus HY9901
2022-07-08 01:43LingZUOLiangchuanCHENShuaiYANGXingXIAOFuyuanZENGJunlinWANGWeijieZHANGHuanyingPANG
Asian Agricultural Research 2022年5期
Ling ZUO, Liangchuan CHEN, Shuai YANG, Xing XIAO, Fuyuan ZENG, Junlin WANG, Weijie ZHANG, Huanying PANG
Fisheries College of Guangdong Ocean University, Zhanjiang 524088, China; Guangdong Provincial Key Laboratory of Pathogenic Biology and Epidemiology for Aquatic Economic Animals, Key Laboratory of Diseases Controlling for Aquatic Economic Animals of Guangdong Higher Education Institutions, Zhanjiang 524088, China
Abstract [Objectives] To clone and analyze the TpiA gene of Vibrio alginolyticus HY9901. [Methods] According to the TpiA gene sequence of V. alginolyticus, a pair of specific primers was designed, and its full length was amplified by PCR. [Results] The full length of TpiA gene is 771 bp, encoding 256 amino acid residues in total, and the NCBI accession number is OM906798. According to the deduced amino acid sequence, its molecular weight was predicted to be about 26.975 48 kDa, and its isoelectric point was 4.78. The amino acid sequence of the N-terminal signal peptide structure was predicted, and it was found that there was no obvious signal peptide cleavage site, no signal peptide, and no transmembrane region; the amino acid sequence contained 3 N-glycosylation sites, 4 protein kinase C phosphorylation sites, 2 casein kinase II phosphorylation sites, 6 N-myristoylation sites, 7 microbody C-terminal target signal site, and 1 triose phosphate isomerase active site. The prediction results of protein subcellular localization showed that TpiA may be located in mitochondria or cytoplasm, with probability of 39.1% and 34.8%, respectively. The amino acid sequence of the TpiA gene of V. alginolyticus shared 98.83%-99.61% homology with other Vibrio species, and it was clustered into the same subfamily with Vibrio parahaemolyticus and had a close relationship. In the secondary structure prediction, the proportions of α-helix, random coil and extended chain were 44.53%, 41.41% and 14.06%, respectively, and the similarity of its tertiary structure model to template 1aw1.1.A was 85.16%. [Conclusions] This study is intended to provide a basis for further research on the role of TpiA gene in the type III secretion system and related research on antibiotic resistance.
Key words Vibrio alginolyticus, Gene amplification, TpiA, Bioinformatics analysis
1 Introduction
In recent years, due to such factors as the expansion of mariculture scale and the deterioration of environmental conditions, the diseases of China’s aquaculture fishes frequently occur[1]. Vibrio, as one of the most common bacterial diseases[2], exists widely in aquaculture, estuaries, and living organisms[3], and has a huge impact on marine life[4-5].Vibrioalginolyticusis a halophilic and mesophilic Gram-negative bacterium[6], and is a main pathogenic bacteria of fish, shrimp, shellfish and other marine aquaculture animals[9]. It is about 1.4-2.6 μm long and 0.5-0.8 μm wide, mostly in the shape of short rod. It has no structure of capsule and spore, but has terminal and pericytic flagella, which is conducive to movement in solid-liquid medium. Its suitable temperature is 17-35 ℃, the pH range is 6.0-9.0, and the NaCl concentration range is 2%-10%[7-8]. It can lead to human food poisoning, diarrhea enteritis, sepsis and many other diseases[10]. The pathogenic mechanism ofV.alginolyticusis complex. After approaching the host by chemotaxis, it adheres to the host cell through adhesin[11], and then damages the tissue and cells of the host cell by means of invasion and proliferation, and finally, it interferes and destroys the physiology and function of the host through toxin-containing metabolites[12]. Pathogenic factors include biofilms, extracellular products, siderophores, lipopolysaccharides, attachment factors,etc.[13], andTpiAmay be involved in the adhesion to host cells[14]. Triose phosphate isomerase encoded byTpiA, which converts glyceraldehyde-3-phosphate to dihydroxyacetone phosphate, is a key step linking glucose metabolism to glycerol and phospholipid metabolism and is essential for energy generation. Removal of theTpiAgene from growth-optimizedE.colistrains may lead to an increase in the content of attack-end molecules (including DNA and RNA) and the highly toxic metabolite glyoxal that disrupts the nucleotide level[15], and affects the tricarboxylic acid cycle of the strain, thereby affecting the growth of the bacterial cell[16]. A counter-intuitive crossover between carbon starvation and inorganic phosphate signaling is revealed in aTpiA-deficient strain, which requires mutations in the inorganic phosphate signaling machinery to alleviate. Studies have revealed thatTpiAplays an important role in the metabolism, virulence and antibiotic resistance ofPseudomonasaeruginosa[17]. The type III secretion system (T3SS) is closely related to the pathogenicity of bacteria and is a highly conserved secretion system ofV.alginolyticus[18-21], and it is a complex transmembrane channel composed of apparatus proteins, transposon proteins, effector proteins, regulatory proteins and molecular chaperones[22-26].P.aeruginosaT3SS is cytotoxic to a variety of cells, while mutations in theTpiAgene can reduce the expression of the T3SS[17]. The advanced 2-D SDS-PAGE technique was used to study the differentially expressed whole protein map ofV.alginolyticusunder the induction of erythromycin, and it was found that the expression of triose phosphate isomerase decreased after induction, showing that the drug resistance mechanism ofV.alginolyticusis closely related to the energy metabolism ofV.alginolyticus[27]. Studies have shown that mutations in theTpiAgene can reduce the resistance ofP.aeruginosato aminoglycoside antibiotics[17].
TpiAis essential for energy generation, but there are few studies on the specific regulatory mechanism ofTpiAinvolvement inV.alginolyticus. Through cloning and analyzing theTpiAgene ofV.alginolyticusHY9901, we intended to provide a basis for further research on the role ofTpiAgene in the type III secretion system and related research on antibiotic resistance.
2 Materials and methods
2.1 Materials
2.1.1Strain.V.alginolyticusstrain HY9901 was obtained by our laboratory through isolation from the diseased red snapper (Eteliscoruscans) in the sea area of Zhanjiang, Guangdong Province[28], and the cloning vector pET-32a was purchased from Takara Bio Incorporation (USA).
2.1.2Main reagents. ExTaq DNA polymerase was purchased from Takara; bacterial genomic DNA extraction kit and DNA gel recovery kit were purchased from Tiangen Biotech (Beijing) Co., Ltd.; other reagents were of imported or domestic analytical reagents. PCR primer synthesis and sequence determination were completed by Sangon Biotech (Shanghai) Co., Ltd. Antibiotic concentration: Ampicillin (Amp) 100 μg/mL.
2.2 Methods
2.2.1Extraction of total DNA fromV.alginolyticusHY9901. TheV.alginolyticusstrain HY9901 was spread on TSA plate, and a single colony was selected and inoculated in TSB (5%NaCl) medium, and cultured at 28 ℃ with shaking for more than 12 h. Took an appropriate amount of bacterial liquid into an EP centrifuge tube, centrifuged at 10 000 rpm for 1 min to collect bacterial cells, extracted genomic DNA in accordance with the kit instructions, and stored at -20 ℃ for later use.
2.2.2Clone ofTpiAgene. A pair of primers were designed according to theTpiAgene sequence ofV.alginolyticus, the upstream primerTpiA-F1 was: ATGCGTCGTCCTGTAGTGATGG, and the downstream primerTpiA-R1 was: TTAAGCTTTT GCTTTAGCAGCTGC. The extracted total DNA ofV.alginolyticusHY9901 was used as the template, and the PCR reaction conditions were: pre-denaturation at 95 ℃ for 3 min; 95 ℃, 30 s; 58 ℃, 30 s; 72 ℃, 50 s; a total of 30 cycles, and then extended at 72 ℃ for 5 min. The PCR products were examined by 1% agarose gel electrophoresis, and then DNA was recovered by cutting the gel with a gel recovery kit.
2.2.3Sequencing of PCR products. According to the operating steps in the instructions, the PCR product was ligated to the pMD18-T vector, thenE.coliDH5α competent cells were then transformed, screened on LB plates containing ampicillin resistance, and positive clones were picked and sent to Guangzhou Biotechnology Center for sequencing.
2.2.4Bioinformatics analysis ofTpiAgene ofV.alginolyticusHY9901. With reference to the method of Pang Huanyingetal.[25], ExPASy (https://web.expasy.org/cgi-bin/protparam/protparam) was used to analyze the physicochemical properties ofV.alginolyticusHY9901TpiA protein, and SignalP 5.0 Server (https://services.healthtech.dtu.dk/service.php?SignalP-5.0) was used to predict the signal peptide structure ofTpiAamino acid sequence. Transmembrane domains predicted by TMHMM Server 2.0 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0), and the distribution of functional sites in amino acid sequences was predicted by SoftBerryPsite (http://linux1.softberry.com/berry.phtml?topic=psite&group =programs&subgroup=proloc). Subcellular localization was predicted using PSORT II Prediction (http://psort.hgc.jp/form2.html); sequence homology alignment and similarity analysis were performed using NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi); amino acid homology alignment analysis was performed using DNAMAN Version 6.0 (Lynnon Biosoft); ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) and ExPASy Proteomics Server (http://ca.expasy.org) were used to deduce amino acid sequence, determine open reading frame (ORF) , molecular weight calculated value (Mw) and theoretical isoelectric point (pI),etc.Protein structural domains were analyzed using InterProScan Sequence Search (http://www.ebi.ac.uk/Tools/InterProScan). The phylogenetic tree was constructed by neighbor-joining method using Clastal 2.0 and MEGA 7.0 software. Modeling was performed using the SWISS-MODEL (http://www.swissmodel.expasy.org/) program of the ExPASy server and analyzed by the 3D structural analysis software PyMOL Viewer.
3 Results and analysis
3.1 Amplification ofTpiAgeneA specific band of about 771 bp was successfully obtained by PCR amplification (Fig.1). Sequencing showed that theTpiAgene contained an open reading frame of 771 bp, encoding 256 amino acids. The gene was imported to GenBank with accession number OM 906798.
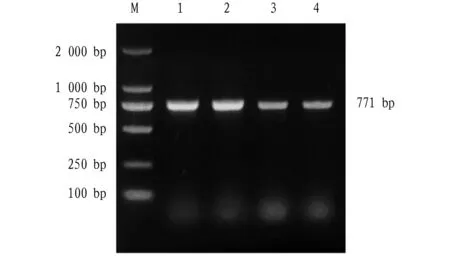
Note: M: DL2000 DNA molecular weight standard; Lane 1-4: TpiA PCR product.Fig.1 Amplification of TpiA gene
3.2 Physicochemical properties ofTpiAWith the aid of ExPASy software, the TpiA protein ofV.alginolyticusHY9901 was analyzed. The results showed that the total number of atoms was 3 788, and the molecular structure was C1190H1891N325O375S7. The theoretical molecular weight was 26.975 48 kDa, and the theoreticalpIvalue was 4.78. The instability coefficient was 24.17 (stable), the fat coefficient was 93.20, the overall average hydrophilicity was -0.016, and the protein overall was hydrophilic. The protein does not contain selenocysteine (Sec) and pyrrolysine (Pyl), and the molar extinction coefficient at the wavelength of 280 nm was 40 005 (mol/cm). The total number of acidic amino acids (Asp+Glu) was 35, the total number of basic amino acids (Arg+Lys) was 22, and the N-terminal was methionine (Met). The half-life of expression in yeast andE.coliwas greater than 20 and 10 h, respectively, and the half-life of expression in mammalian reticulocytesinvitrowas estimated to be 30 h.
3.3 Sequence analysis ofTpiAThe N-terminal signal peptide structure ofTpiAamino acid sequence was predicted using SignalP 5.0 Server program, it is found that there is no obvious signal peptide cleavage site and no signal peptide. The protein was predicted to have no transmembrane domain by TMHMM Server 2.0 program. Through prediction using SoftBerry-Psite program, it is found that amino acid sequence contained 3 N-glycosylation sites (13-16aa, 71-74aa, 105-108aa), 4 protein kinase C phosphorylation sites (98-100aa, 176-178aa) , 191-193aa, 214-216aa), 2 casein kinase II phosphorylation sites (181-184aa, 225-228aa), 6 N-myristoylation sites (35-40aa, 64-69aa, 74 -79aa, 163-168aa, 175-180aa, 231-236aa), 7 microbody C-terminal target signal sites (15-17aa, 59-61aa, 91-93aa, 177-179aa, 248-250aa, 252-254aa, 254-256aa), 1 triose phosphate isomerase active site (167-177aa), as shown in Fig.2. Using PSORT II Prediction to predict protein subcellular localization, the results show thatTpiAmay be located in mitochondria or cytoplasm, 39.1% and 34.8%, respectively, followed by cytoplasm and nucleus, with a probability of 8.7%, while the probability of being located in peroxidase and vacuole is 4.3%.
3.4 Homology and evolution analysis of TpiAThrough BLAST analysis, it found that the TpiA ofV.alginolyticushad high homology with the TpiA of other Vibrio species. Among them, the homology with the TpiA amino acid sequence ofV.parahaemolyticuswas up to 99%, and the multiple sequence similarity comparison shows thatTpiAinVibriowas highly conserved (Fig.3).
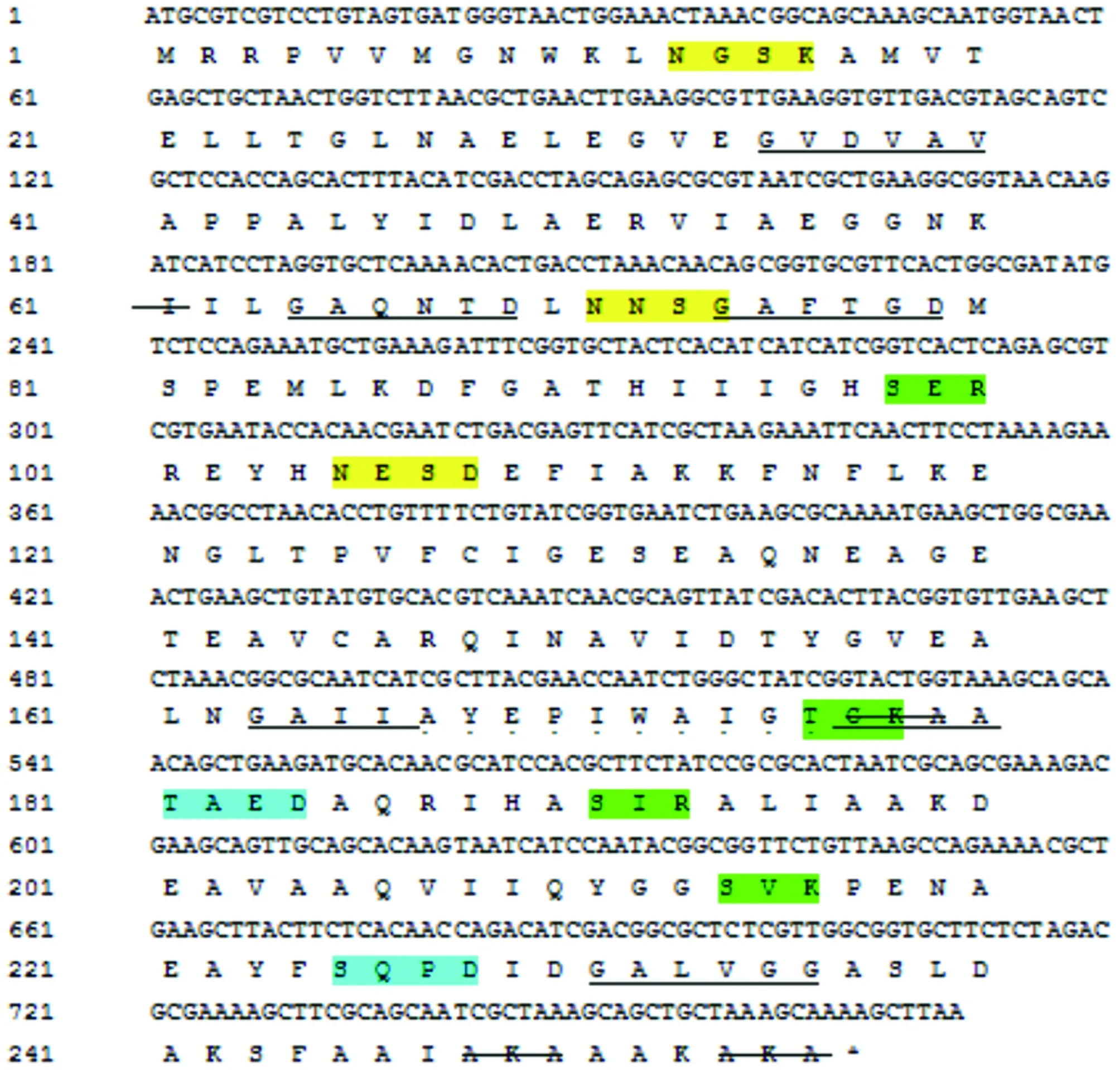
Note: terminators are indicated by *, N-glycosylation sites are indicated in yellow, protein kinase C phosphorylation sites are indicated in green, casein kinase II phosphorylation sites are indicated in blue, N-myristoylation sites are indicated by black underline, the microbody C-terminal target signal site is indicated by strikethrough, and the triose phosphate isomerase active site is indicated by underlined dots.Fig.2 TpiA gene nucleotide and its encoded amino acid sequence

Note: V. alginolyticus (OM906798); V. harveyi group (WP_010445251.1); V. owensii (WP_199482583.1); V. parahaemolyticus (WP_025501099.1); V. rotiferianus (WP_171356767.1).Fig.3 Homology comparison of the deduced amino acid sequence of TpiA gene
With the aid of MEGA 7.0 software, we constructed a phylogenetic tree by combining the deduced amino acid sequence of TpiA with other microorganisms using Neighbor-joining method. The results show that the TpiA protein ofV.alginolyticusHY9901 was obviously the same subfamily asV.parahaemolyticus, indicating that they were closely related (Fig.4), which is consistent with the traditional classification results of morphological and biochemical characteristics.

Fig.4 TpiA amino acid phylogenetic tree constructed based on NJ method
3.5 Functional domain, secondary and tertiary structure prediction of TpiAThrough prediction using the SMART program, it was found that there is a TpiA functional domain (4-248aa) (Fig.5). In the secondary structure prediction, alpha helix accounted for 44.53%; random coil accounted for 41.41%; the extended strand accounted for 14.06% (Fig.6).

Fig.5 Functional domains of TpiA
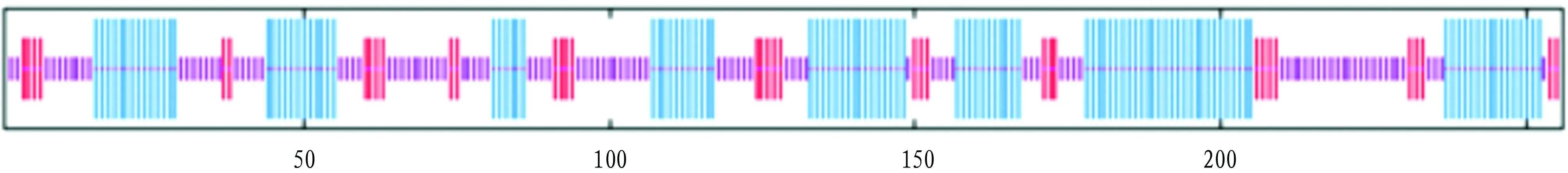
Note: blue: alpha helix; purple: random coil; red: extended strand.Fig.6 Secondary structure prediction of TpiA
The amino acid sequence of TpiA was imported to the SWISS-MODEL program, and homologous proteins were automatically searched as templates to obtain the tertiary structure model of TpiA single subunit (Fig.7).
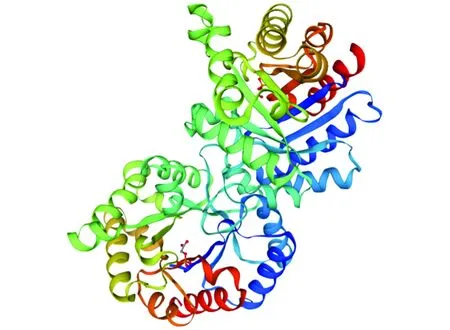
Note: template: 1aw1.1.A; similarity: 85.16%.Fig.7 Tertiary structure prediction of TpiA
3.6 TpiA protein network interactionIn the protein network interaction, it can be found that the TpiA protein can be co-expressed with pgk, fbaA, pgi, epd, VMC-15210, gapA, eno, VMC-19260, VMC-19270, and there are pgk, fbaA, pgi, eno in Text mining (Fig.8).
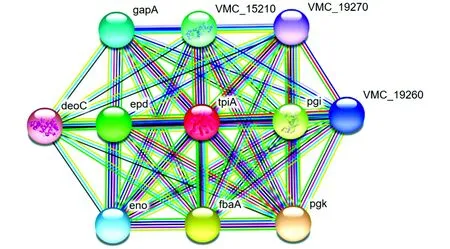
Fig.8 TpiA protein network interaction
4 Discussion
Based on the amino acid sequence of theTpiAgene ofV.alginolyticusHY9901, through a comprehensive comparative analysis of its secondary structure and tertiary structure, transmembrane region, hydrophilicity, protein network interaction and other parameters, we predicted the possible role of TpiA protein. Triose-
phosphate isomerase (TPI) is a catalytic enzyme in the process of glucose metabolism, and it can catalyze the conversion of dihydroxyacetone phosphate (DHAP) to glyceraldehyde 3-phosphate (G3P). Glycolytic pathway can be associated with lipid metabolism, pentose phosphate pathway, gluconeogenesis pathway through DHAP and G3P. Therefore, TPI plays an important role in regulating the metabolic balance of the body[28]. At present, there are few studies on theTpiAgene and protein inVibrio. InP.aeruginosa, Crc, Hfq, Small RNA (including miRNA, ncRNA, siRNA, snoRNA, piRNA, rasiRNA,etc.) and CrcZ are central regulators of carbon metabolism. Through screening for mutants of carbon metabolism-related genes, we found that mutations in theTpiAgene reduce the expression of the type III secretion system (T3SS) and bacterial resistance to aminoglycoside antibiotics.TpiAis a triose phosphate isomerase that converts glyceraldehyde-3-phosphate to dihydroxyacetone phosphate, which is a key step linking glucose metabolism to glycerol and phospholipid metabolism. The mutatedTpiAgene enhances bacterial carbon metabolism, respiration, and oxidative phosphorylation, increases membrane potential, and promotes the intake of aminoglycoside antibiotics. Further studies showed that the level of CrcZ was increased in mutatedTpiAdue to enhanced stability. In the context of mutatedTpiA, mutation of the crcZ gene restores T3SS gene expression and bacterial resistance to aminoglycoside antibiotics. This study revealed thatTpiAplays a great role in metabolism, virulence and antibiotic resistance ofP.aeruginosa[17]. Pyruvaldehyde is a highly toxic metabolite produced in all organisms and it can attack terminal molecules (including DNA and RNA) and disrupt nucleotide levels. Deletion of theTpiAgene from growth-optimizedE.colistrains resulted in increased levels of pyruvaldehyde. In aTpiA-deficient strain, it revealed a counterintuitive crossover between carbon starvation and inorganic phosphate signaling, which requires mutations in the inorganic phosphate signaling machinery to alleviate. Split flux of glycolytic intermediates through low glycolytic depletion requires a large number of synchronized and coordinated mutations at non-intuitive network locations to retune metabolic flux for optimal growth[15]. Such mutations include systematic inactivation of the phosphotransferase system (PTS) and alteration of the activity of all biosynthetic enzymes in the cell. As an exocrine protein of Toxoplasma gondii, triose phosphate isomerase can trigger a strong immune response in the host[29].
5 Conclusions
In this study, we successfully amplified the complete gene sequence ofTpiAfromV.alginolyticusHY9901. The full length ofTpiAgene is 771 bp, encoding 256 amino acid residues in total. Its protein is stable and hydrophilic, and its molecular weight was predicted to be about 26.975 48 kDa, and its isoelectric point is 4.78. There is no obvious signal peptide cleavage site, no signal peptide, and no transmembrane region; the amino acid sequence containes 3 N-glycosylation sites, 4 protein kinase C phosphorylation sites, 2 casein kinase II phosphorylation sites, 6 N-myristoylation sites, 7 microbody C-terminal target signal site, and 1 triose phosphate isomerase active site. The prediction results of protein subcellular localization showed thatTpiAmay be located in mitochondria or cytoplasm, with probability of 39.1% and 34.8%, respectively. The amino acid sequence of theTpiAgene ofV.alginolyticusshared 98.83%-99.61% homology with otherVibriospecies, and it was clustered into the same subfamily withV.parahaemolyticusand had a close relationship. In the secondary structure prediction, the proportions of α-helix, random coil and extended chain were 44.53%, 41.41% and 14.06%, respectively, and the similarity of its tertiary structure model to template 1aw1.1.A was 85.16%. In the protein network interaction, it can be found that the TpiA protein can be co-expressed with pgk, fbaA, pgi, epd, VMC-15210, gapA, eno, VMC-19260, and VMC-19270. It is feasible to further study the role and mechanism of this gene in transcriptional regulation through knocking out theTpiAgene inV.alginolyticusand comparing with the wild strains.
 Asian Agricultural Research2022年5期
Asian Agricultural Research2022年5期
- Asian Agricultural Research的其它文章
- Status Investigation and Countermeasures of Green Development of Black Talc Industry Chain in Guangfeng District, Jiangxi Province
- Inhibitory Effect of Lactic Acid Bacteria on Salmonella
- Antagonistic Effects of Sphingomonas and Pseudomonas aeruginosa on 4 Kinds of Pathogenic Bacteria of Ginseng
- Bioinformatics Analysis of Xylanase xynMF13-GH10 Gene from Mangrove Fungi
- Methodological Research in the Field of Civil Aviation Safety
- Safety Evaluation of Water Environment Carrying Capacity of Five Cities in Ningxia Based on Ecological Footprint of Water Resources