
成人散发型神经元包涵体病临床分析
2022-07-04 08:22陆梦如梅珊珊许二赫
中国实用神经疾病杂志 2022年4期
陆梦如 梅珊珊 武 霄 许二赫
1)广西医科大学第二附属医院,广西 南宁 530027 2)首都医科大学宣武医院,北京 100053
神经元核内包涵体病(neuronal intranuclear inclusion disease,NIID)是一种进展缓慢的神经系统退行性疾病,其病理特征是在中枢和外周神经系统及其他组织中存在嗜酸性泛素阳性和p62 阳性的核内包涵体[1-2]。临床表现极为异质,家族性和散发性病例均有报道。不同的NIID家族具有来自多个系统的不同表型,即使在同一个家系中,不同成员的表型可以有差异。有时仅根据临床表现很难诊断NIID,以特发性震颤、头痛为首发症状的临床罕见。本研究报道6 例NIID 并结合文献复习,提高临床医生对该病的认识。
1 对象和方法
收集首都医科大学宣武医院神经内科2020-09—2021-12 收治的6 例诊断的成年散发型NIID 患者的临床资料。回顾性分析6例患者的临床表现、实验室检查、影像学特点、皮肤病理、NOTCH2NLC基因。所有患者均常规行血常规、血生化、糖化血红蛋白、梅毒、艾滋、肿瘤指标物、自身免疫性抗体等实验室检查。6 例均行头颅MRI、脑电图、肌电图检查,5 例行皮肤病理活检,1 例行脑活检,6 例均行NOTCH2NLC基因GGC测序、腰穿脑脊液检测。6例均完成简易精神状态量表(mini-mental state examination,MMSE)、蒙特利尔认知评估(Montreal cognitive assessment,MoCA)。
2 结果
2.1 临床特点6 例患者均为成年起病,病例1、病例2 有糖尿病史多年,病例1 以发作性脑病、认知障碍为核心表现,伴泌尿系统、内分泌系统、呼吸系统、周围神经损害。病例6 以震颤为首发表现,认知正常。病例3以反复发作性头痛为核心症状,发作性脑病表现,伴进行性认知功能损害。6例患者头颅DWI均见双侧额颞叶顶叶皮髓交界区线样高信号,2例脑脊液蛋白水平轻微升高,5例皮肤病理检查均可见神经元核内包涵体。5例患者神经传导速度异常,尤其是四肢运动和(或)感觉神经传导速度减慢;6例长读长基因组测序NOTCH2NLC 基因的GGC 重复扩增,均>60。见表1。
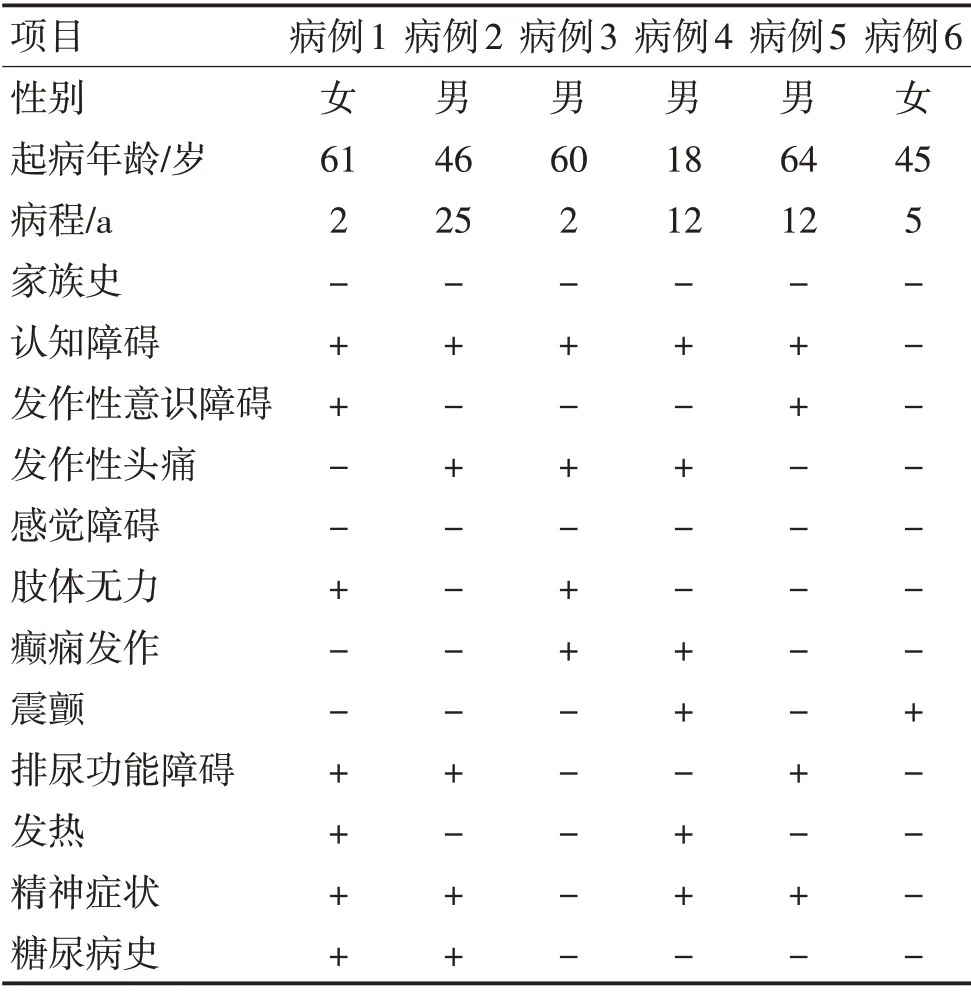
表1 6例NIID患者的临床表现Table 1 Clinical manifestations of 6 NIID patients
2.2 实验室检查6例患者血生化、糖化血红蛋白、梅毒、艾滋、肿瘤指标物、自身免疫性抗体、副肿瘤抗体、自身免疫性脑炎抗体均在正常范围内,均行腰穿检查,脑脊液常规均正常,2例脑脊液蛋白轻度升高,2例脑脊液IgG OB阳性(表2)。
2.3 影像学检查6例头颅MRI检查均有明显脑白质病变,基底节区腔隙灶(图1A)。其中5例DWI有飘带征(图1B~F),4例可见脑萎缩。病例3见右侧颞枕顶叶脑回肿胀(图1C)。病例6 FDG-PETCT 检查可见双侧壳核葡萄糖代谢增高,双侧上顶叶、额顶叶交界区代谢中度减低,左侧额颞叶交界区、左侧下颞叶、左侧枕叶代谢轻中度减低(图2)。见表2。
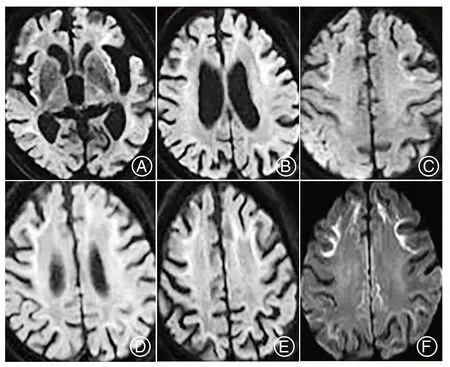
图1 A:病例1脑内多发缺血灶、梗死灶,脑白质变性,脑萎缩;B:病例2双侧额颞叶皮髓交界区可见线样高信号;C:病例3 脑白质异常信号,右侧额颞枕脑回样肿胀,双侧额颞叶顶叶皮髓交界区可见线样高信号;D、E、F:分别为病例4、病例5、病例6 DWI上高信号Figure 1A:Multiple ischemic foci,infarct foci,white matter degeneration,and brain atrophy in the brain of case 1;B:Line-like high signal in the bilateral frontotemporal cortex-medullary junction in case 2;C:Abnormal signal of white matter,right frontotemporal occipital gyri-like swelling,line-like high signal in the bilateral frontotemporal parietal cortex-medullary junction in case 3;D,E,F:DWI of case 4,case 5,and case 6,respective high signal

图2 病例3头颅FDG-PETCT示,双侧上顶叶、额顶叶交界区代谢中度减低;左侧额颞叶交界区、左侧下颞叶、左侧枕叶代谢轻中度减低Figure 2 FDG-PETCT of the head of case 3 showed that the metabolism of the bilateral upper parietal lobe and the fronto-parietal junction was moderately decreased;the left frontotemporal junction,the left inferior temporal lobe,and the left occipital lobe were mildly to moderately decreased

表2 6例NIID患者的辅助检查结果Table 2 Auxiliary examination results of 6 NIID patients
2.4 病理检查5例皮肤病理显示皮肤表皮无明显异常,真皮层胶原纤维增生,个别汗腺导管上皮细胞内可见核内包涵体(图3)。1例腓肠神经病理提示有髓鞘神经纤维丢失,不典型洋葱球,符合髓鞘周围神经病理改变特点。1 例脑组织活检提示个别神经元及胶质细胞核内ubiquitin 及P62 阳性的小圆形结构(图4)。
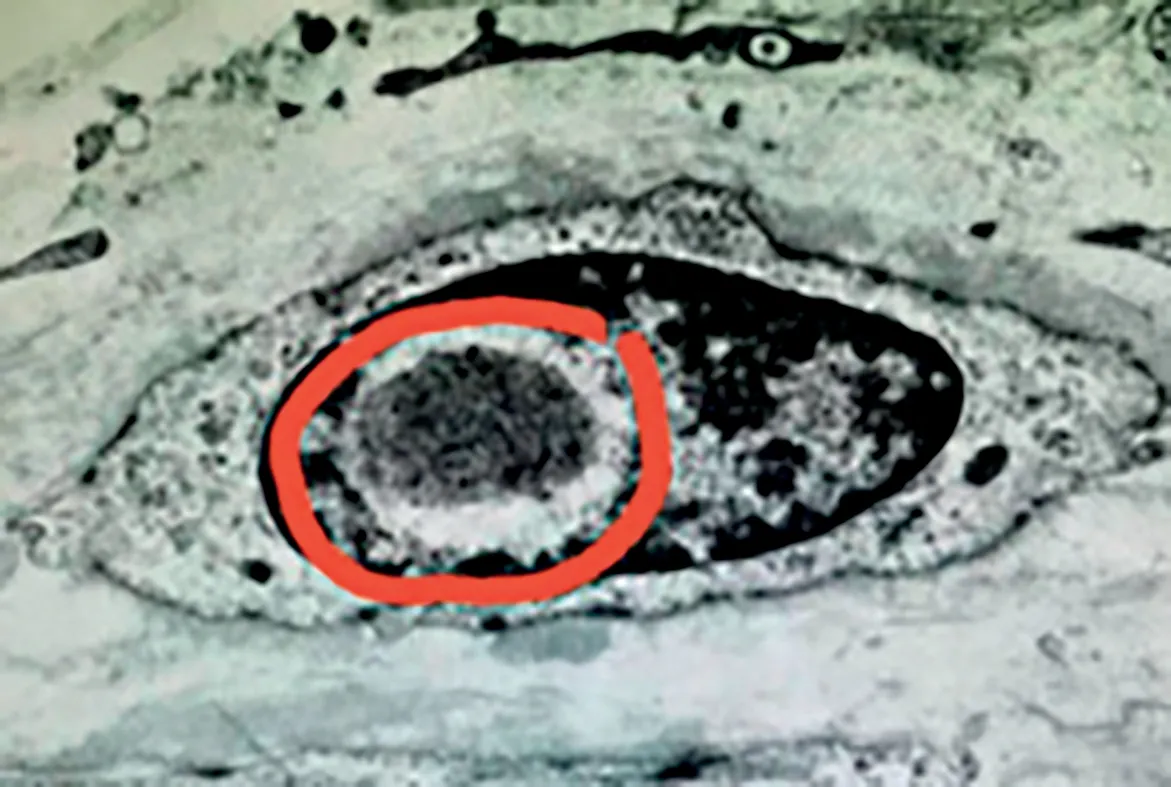
图3 病例1汗腺导管上皮细胞内可见核内包涵体(红色圆框所示)Figure 3 Intranuclear inclusions(indicated by the red circle)were seen in the ductal epithelial cells of the sweat glands in case 1
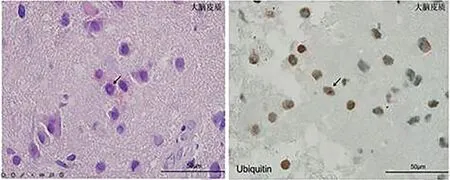
图4 病例3脑组织病理切片可见个别神经元及胶质细胞内Ubiguitin及P62阳性的小圆或小环形结构Figure 4 The pathological section of the brain tissue of case 3 showed small round or small ring structures positive for Ubiguitin and P62 in individual neurons and glial cells
2.5 基因检测6 例基因组测序NOTCH2NLC 基因的GGC 重复扩增,其中病例6 GGC 重复次数为60,其余病例NOTCH2NLC 基因的GGC 重复扩增均为100次以上(图5)。

图5 A、B:分别为病例1 病例6 NOTCH2NLC 基因GGC 检测,NOTCH2NLC 基因5’端非翻译区进行重复引物聚合酶链反应扩增结果[横坐标:碱基对(bp);纵坐标:信号强度]Figure 5 A and B are GGC detection of NOTCH2NLC gene in case 1 and case 6,respectively.The results of repeated primer polymerase chain reaction amplification of the 5’-end untranslated region of NOTCH2NLC gene(abscissa:base pair(bp);ordinate:signal)
2.6 治疗所有病例给予对症治疗,疗效欠佳。病例3 给予静脉输注甲强龙治疗,500 mg,qd,3 d;250 mg,qd,3 d。后改泼尼松60 mg口服,逐渐减量,其间予辅酶Q10、丁苯酞营养神经,病情好转出院。
3 讨论
NIID 于1968 年由LINDENBERG 等[3]首次报道,之后相关病例仅有零星报道。2011年后,SONE首次在NIID 患者的皮肤活检中发现泛素阳性包涵体,此后,经皮肤活检确诊的NIID 病例数迅速增加。2019年,来自不同研究组的研究人员报告了家族性和散发性亚洲NIID患者NOTCH2NLC基因的GGC重复扩增,提示NOTCH2NLC可能与该病的发病机制有关[4-7]。到目前为止,NIID 的潜在机制尚不清楚。目前尚无NOTCH2NLC 基因检测与皮肤活检诊断价值的对比研究。
本研究6例患者,男4例,女2例,均为散发病例,平均发病年龄42.75 岁。6 例均累及神经系统,其中病例1和病例5以认知障碍为核心表型,病例2、病例3、病例4均以发作性头痛为核心表型,病例6以震颤为核心表型。以认知障碍的2例平均年龄62.5岁,以头痛起病的3例患者起病年龄18~60岁,与既往文献报道成人散发型NIID 累及神经系统的发病年龄一致。病例1和病例2有糖尿病。4例患者伴排尿功能障碍,所有患者均无感觉障碍,5 例神经传导速度异常,1 例未能完成该项检查。5 例患者脑电图均有不同程度慢波表现。此外,2例患者有发热,4例有精神症状,2 例有癫痫发作。病例1 反复泌尿系感染、尿潴留、反复发热,首次入住泌尿外科,后因血糖升高入住内分泌科,查头颅DWI提示额颞交界高信号,最后通过基因、皮肤病理明确诊断。病例5反复发作性晕厥、言语困难,多次诊断为短暂性脑缺血发作,初期影像检查不典型,本次入院查头颅DWI 额颞叶显示高信号影。痴呆、发作性脑病是NIID 最常见的临床表现,但发病早期可能会合并自主神经系统、泌尿系统、内分泌系统相关症状,甚至患者首次就诊的主要原因,导致早期诊断困难。本研究中病例2、3、4以反复发作性头痛为核心表现,病程2~25 a,频率逐渐增多,伴进行性认知功能下降。病例4伴发热、震颤、精神症状,通过文献检索,国内有1例以头痛为首发症状的NIID 报道[8]。目前研究报道不明原因的头痛为NIID 的一种新的早期表型。LIANG等[9]报告1例35岁女性NIID患者,首发症状为先兆偏头痛,从早期孤立的偏头痛发作到后期伴发作性脑病意识障碍的偏头痛发作,提出一种疾病在不同阶段的连贯过程,然而发病起始因素仍不清楚。WANG 等[10]报道1 例首发症状为偏头痛的患者,起病年龄8岁,逐渐进展,该患者的头颅MRI 提示右侧大脑半球广泛水肿,灌注成像提示右侧大脑半球缺血性改变,与本研究病例3头颅MRI示右侧颞叶水肿相似。WANG 等[10]基于他们的临床数据提出了两种可能性:第一种是脑灌注过低和葡萄糖代谢过低可能通过激活神经退行性变的致病通路,促进嗜酸性包涵体在神经元中的积累,进而导致NIID的进展;第二种是嗜酸性内含物的积累首先发生,通过改变神经元的氧需求而导致脑血管功能障碍的发展,导致偏头痛。本研究3 例头痛患者均未行脑灌注成像检查,无法判断是否与脑灌注具有相关性。此外,由于头痛是一种常见的临床疾病,不能完全排除头痛和NIID作为两种独立的临床条件存在的可能性,需要更多的病例和进一步的研究探索这两种疾病之间的关系。本研究病例6 以肢体不对称性震颤为主要表现,逐渐进展,结合该患者病史,很容易误诊为PD 或特发性震颤(essential tremor,ET)。但该患者头颅DWI 累及皮质髓质交界处的带状征影像学表现,伴脑萎缩,符合典型的NIID。FDG-PETCT的双侧上顶叶、额顶叶交界区代谢中度减低与病灶相匹配。长读长基因组测序提示,NOTCH2NLC基因的GGC 重复扩增。该患者认知正常,长期服用抗震颤药物治疗无效。国内以震颤为首发表现的NIID报道甚少。最近的一项研究也报道了一些家族性NIID患者最初表现为特发性震颤样症状,随后出现其他明显的神经系统症状和典型的MRI改变[11]。目前研究[12]表明,在PD 表型的NIID 患者中,GGC 重复扩张中有GGA 和AGC 两种形式的重复中断。在PD 中GGC扩张中没有GGA中断,而约12%的NIID患者存在GGA中断。此外,PD患者AGC中断的频率至少比NIID患者高3倍。
NIID的发病年龄差异很大,根据发病年龄可分为3个亚组:婴儿型、少年型和成人型。成人发病的临床表现多种多样,包括痴呆、帕金森病、震颤、小脑共济失调、癫痫、周围神经病变、自主神经症状和视网膜病变。基于NIID 的神经症状,TIAN 等[13]将家族病例受试者分为3个亚组:肌无力型NIID-M、帕金森病型NIID-P和痴呆型NIID-D。3 个亚组NOTCH2NLC 基因中GGC的重复数分别 为 118~517、66~102和91~268个单位,而健康组的重复数为5~38个单位,但未观察到 GGC 大小与NIID发病严重程度或年龄之间的相关性。本研究报告的6 例与文献一致。头颅弥散加权成像在皮质髓质交界处的对称高信号具有重要诊断价值[14-16],据报道,具有家族史的成人起病型NIID患者头颅MRI上述特征阳性率仅为37.5%,一些散发病例不具有此类影像学特征[17-20]。
CHEN 等[21]首次系统总结了经基因检测证实的51例NIID患者的临床症状。NIID在不同系统中的初始症状百分比分别为呼吸系统37.3%,神经系统21.6%,消化系统13.7%,泌尿系统11.8%,循环系统7.8%,生殖系统3.9%,运动系统2.0%;不同系统中位发病年龄分别为运动系统3 岁,生殖系统28.5 岁,消化系统30岁,循环系统38.5岁,呼吸系统42岁,神经系统50岁,泌尿系统55岁。关节疼痛、肢体无力或疲劳等运动系统症状可以是NIID 患者最早的初始症状。因此,临床应更多关注其他系统的早期症状,并早期进行基因检测、头颅影像学检查、皮肤病理明确诊断。此外,NOTCH2NLC基因的GGC扩增与神经系统变性病、周围神经病存在一定重叠。国际上也有学者倾向将NIID 定义为一类谱系疾病(NIID related disorders,NIIDRD)。
NIID 的临床特征具有高度异质性,早期诊断困难,痴呆和阵发性脑病是常见的突出表型,以震颤为主的NIID 容易被误诊为ET。对于一些不明原因反复头痛的患者,DWI 皮髓质交界区高信号具有重要提示价值,DWI阴性不能排除NIID,尤其仅累及外周或自主神经系统的NIID,对动态影像改变的长期随访可能有助于诊断。当患者无法行皮肤活检时,基因检测有参考价值。本病目前无特异性治疗,以支持、对症治疗为主。
猜你喜欢
保健与生活(2021年21期)2021-11-11
健康大视野(2020年7期)2020-04-26
家庭科学·新健康(2019年9期)2019-10-21
养生大世界(2018年10期)2018-10-18
东坡赤壁诗词(2018年3期)2018-07-16
意林·少年版(2018年24期)2018-01-05
健康女性(2016年11期)2017-02-14
扬子江(2016年1期)2016-05-19
经营者·汽车消费报告(2013年11期)2014-01-23
美文(2009年10期)2009-06-04
