
几种新型3LCuCl配合物的合成及其对甲醇氧化羰基化反应催化性能研究
2022-07-02 05:08王云飞刘昌盛杨喜庆胡泉源
湖北大学学报(自然科学版) 2022年4期
王云飞,刘昌盛,杨喜庆,胡泉源
(湖北大学化学化工学院, 湖北 武汉 430062)
0 引言
现代化工生产的许多过程都产生大量的一氧化碳(CO),例如合成氨原料气、黄磷生产尾气以及钢铁工业的高炉气和转炉气,对CO气体的转化利用是目前C1化学的重要任务,合成碳酸二甲酯(DMC)是CO转化方向之一. DMC是绿色化工产品,可作为无毒有机溶剂使用,它是聚碳酸酯(PC)的主要原料,也可以在精细有机化工生产中取代剧毒的光气和硫酸二甲酯用作甲基化和羰基化剂[1-3]. 利用甲醇氧化羰基化法合成DMC在工业界已获得广泛运用[4-7],其技术核心是铜系催化剂的使用,由于国外技术封锁,国内目前唯一工业催化剂由成都有机所提供,尚存在价格昂贵、催化寿命短的问题. 为此,我们对开发新型甲醇氧化羰基化催化剂进行了探索.
反应原理如下:
其催化原理基于Cu(I)/Cu(II)的循环转化,氧气的活化利用效果对催化剂性能的体现极为重要,我们以此为出发点进行催化剂的设计. 文献显示节肢动物的血蓝蛋白酶能把氧气从体外运输到体内,有效活化氧气[8-9]参与生理代谢,其活性中心是3个4-甲基咪唑与亚铜离子的配合物,配位方式如图1.
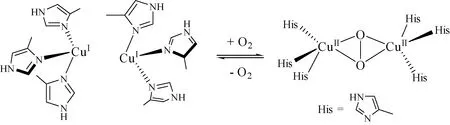
图1 血蓝蛋白酶活化O2的过程
受血蓝蛋白酶结构启发,结合工业反应条件(120 ℃,2.4 MPa),我们拟设计合成具有以下结构特点的催化剂(如图2). 3种催化剂(3LNCuCl,3LoCuCl,3LSCuCl)均为螯合物结构,以期提高其在高温高压下的稳定性,并通过N-烷基化和O-烷基化引入两个长碳链的正己基以增加催化剂的溶解性. 为了了解配位强度对催化剂性能的影响,分别以N-N-N型配体(3LN)、N-O-N型配体(3Lo)和N-S-N型配体(3LS)的方式与氯化亚铜形成配位.
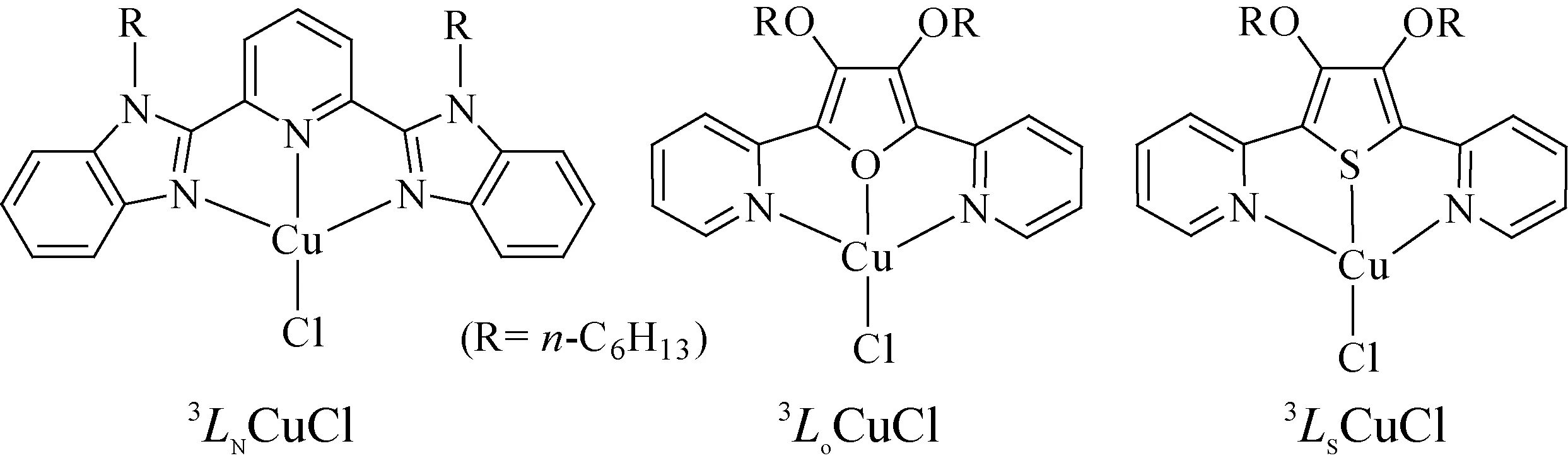
图2 三种合成催化剂结构
1 实验部分
1.1 实验仪器与试剂主要仪器: 安徽科幂NSG通用型高压反应釜,福立气相色谱仪(GC9790plus), 集热式搅拌器.主要试剂: 邻苯二胺、 2,6-吡啶二甲酸、 溴己烷、2-氯甲基吡啶、2-吡啶甲醇、草酸二乙酯、氢化钠.
1.2 2,6-双(N-己基-苯并咪唑基)吡啶(3LN)的合成将无水DMF 150 mL、2,6-双苯并咪唑基吡啶6.22 g (0.02 mol)、NaH 1.20 g(0.05 mol)依次加入250 mL的三口烧瓶中,在氮气保护下搅拌至不再有气体产生,升温至80 ℃,缓慢滴加1-溴己烷8.25 g (0.05 mol),反应5 h. 过滤,滤液浓缩至10 mL,加入乙酸乙酯100 mL,并用10%的NH4Cl溶液洗涤,有机相用无水硫酸镁干燥过夜,柱层析分离纯化(硅胶,流动相VEA∶VPE=3∶4),得淡黄色油状产物7.59 g,产率79.2%.1H NMR(400 MHz, CDCl3)δ: 8.34(d,J=12 Hz, 2H), 8.08(t,J=12 Hz, 1H), 7.90(d,J=12 Hz, 2H), 7.48(d,J=12 Hz, 2H), 7.37(m, 4H), 4.71(t,J=7.2 Hz, 4H), 1.72(m, 4H), 1.07(m, 12H), 0.64(t,J=7.2 Hz, 6H).13C{1H} NMR (100 MHz, CDCl3)δ:150.07, 142.85, 138.13, 136.29, 125.46, 123.48, 122.71, 120.33, 110.35, 44.89, 31.16, 30.01, 26.32, 22.36, 13.75. MS: C31H37N5=479.672 0 (Theo.), 480.307 9 (M+H, Found).
1.33LNCuCl的合成氮气保护下将含氯化亚铜1.98 g (0.01 mol)的乙腈溶液50 mL加热至80 ℃,缓慢滴入3LN配体2.40 g (0.005 mol)的乙腈溶液10 mL,搅拌反应4 h,溶液变为绿色,冷却析晶,减压过滤,滤饼用冰乙腈洗涤,真空干燥,得绿色固体2.21 g,收率76.5%. MS: C31H37N5CuCl=578.668 0 (Theo.), 579.196 0(M+H, Found).
1.4 2-吡啶甲基醚的合成将2-吡啶甲醇3.27 g(0.03 mol)、2-氯甲基吡啶盐酸盐4.92 g(0.03 mol)和无水 1,4-二氧六环80 mL分别加入烧瓶中,在搅拌下慢慢加入1.68 g (0.07 mol)的氢化钠,室温下充分反应4 h. 过滤,滤液浓缩得油状物,柱层析分离(硅胶,VEA∶VPE=3∶7)得到5.35 g 产物,产率约为89.1%.1H NMR (400 MHz, CDCl3)δ: 8.48(m, 2H), 7.62(m, 2H), 7.44(d,J=7.2 Hz, 2H), 7.11(m, 2H), 4.71(s, 4H).
1.5 2,5-二(2-吡啶基)-3,4-二羟基呋喃的合成将二(2-吡啶甲基)醚4.00 g(0.02 mol)、草酸二乙酯2.92 g(0.02 mol)和无水THF 100 mL迅速加入干燥的500 mL三口烧瓶中,把烧瓶放入冷阱中,氮气保护下搅拌,待其液体温度降到-35 ℃,用玻璃注射器注入浓度为2.0 mol/L LDA溶液50 mL. 在低温恒温下搅拌5 h,慢慢反应液回复至室温,继续搅拌1 h. 加入少量乙醇到反应液中猝灭过量的LDA,将反应液减压浓缩,得到固体粉末. 往其加入100 mL的水,并缓慢滴加盐酸中和至pH为7,有沉淀产生. 过滤,粗产物用甲苯重结晶,得到棕色固体1.64 g,产率32.3%.1H NMR (400 MHz, CDCl3)δ:8.44(m, 2H), 7.74(m,2H), 7.53(m,2H), 7.07(m, 2H), 6.65(br, 1H).13C{1H} NMR (100 MHz, CDCl3):151.378, 147.912, 140.122, 137.125, 132.462, 120.197, 115.865.
1.6 2,5-二(2-吡啶基)-3,4-二己氧基呋喃(3Lo)的合成合成方法参照1.2,柱层析分离(硅胶,VEA∶VPE=1∶9),得到无色油状物,产率81.1%.1H NMR (400 MHz, CDCl3)δ: 8.77(m, 2H), 8.13 (m, 2H), 7.84(m, 2H), 7.47(m, 2H), 4.42(t,J=7.2 Hz, 4H), 1.82 (m, 4H), 1.47 (m, 4H), 1.30(m, 8H), 0.90(t,J=7.2 Hz, 6H).13C{1H} NMR (100 MHz, CDCl3):165.295, 149.904, 148.332, 136.959, 126.776, 125.087, 66.126, 31.456, 28.652, 25.577, 22.530, 14.002. MS: C26H34N2O3=422.256 9(Theo.), 423.248 5 (M+H, Found).
1.73LoCuCl的合成制备方法参照1.3,得到浅绿色固体产物,收率94.8%.MS: C26H34N2O3CuCl+CH3OH=552.1811(Theo.), 552.230 7 (Found).
1.8 2,5-二(2-吡啶基)-3,4-二羟基噻吩的合成合成方法参照1.5,固体用甲苯和石油醚混合溶剂重结晶,得棕色固体1.64 g,产率30.3%.1H NMR (400 MHz, CDCl3)δ:12.67(s, 1H),8.50 (m, 2H), 7.76(m, 2H), 7.32(m, 2H), 7.16(m, 2H).13C{1H} NMR (100 MHz, CDCl3):154.585, 149.256, 146.966, 137.522, 120.660, 118.292, 109.597.
1.9 2,5-二(2-吡啶基)-3,4-二己氧基噻吩(3LN)的合成合成方法参照1.2,产率为73.1%.1H NMR (400 MHz, CDCl3)δ:8.50 (m, 2H), 8.01 (m, 2H), 7.62 (m, 2H), 7.05(m, 2H), 4.06(t,J=7.2 Hz, 4H), 1.74(m, 4H), 1.40(m, 4H), 1.26(m, 8H), 0.83(t,J=7.2 Hz, 6H).13C{1H} NMR (100 MHz, CDCl3): 151.690, 149.358, 148.094, 136.292, 127.875, 121.709, 120.811, 73.461, 31.636, 30.205, 25.759, 22.646, 14.044.
1.103LSCuCl的合成制备方法参照1.3,得到浅绿色固体产物,收率89.4%. MS:C26H34N2O2SCuCl=536.132 0(Theo.), 536.131 4(Found).
1.11 甲醇氧化羰基化催化实验取催化剂0.5 g、甲醇50 mL加入100 mL高压反应釜,密封,按分压2∶1充入CO和O2至1.8 MPa,搅拌并升温至120 ℃,此时总压力达到2.4 MPa,反应过程中尽量保持压力不变. 每间隔1 h取反应中间样品0.5 mL,用气相色谱监测其成分,并按归一法分别折算成DMC、DMM和MA相对含量,计算DMC的时空转化率及其选择性. 气相色谱分离条件:柱箱25 ℃,进样器和检测器温度均为200 ℃,载气流量:1.5 mL/min、尾吹流量:30 mL/min、氢气流量:30 mL/min、空气流量:300 mL/min.
2 结果与讨论
2.1 配合物3LNCuCl的合成原料2,6-双苯并咪唑基吡啶参照文献[10-11]合成,因其分子结构刚性较强,在一般溶剂中溶解度较差,选择DMF作为反应溶剂比较合适. 配合物的合成方法见图3. N-烷基化时,NaH不宜过量太多,为防止DMF分解,反应初期应在室温条件下进行. 在配合物制备时需用新鲜氯化亚铜,并且氮气保护,不然产物的XPS谱图中会出现明显的Cu2+信号.

图3 配合物3LNCuCl的合成方案
2.2 配合物3LoCuCl的合成双吡啶甲醚的合成参照文献[12]改进,溶剂以二氧六环代替乙腈,以氢化钠代替氢氧化钾为碱,在室温条件下反应即可,避免了氯甲基的水解反应,收率由46%显著提高至89%. 对于它与草酸二乙酯的关环反应(见图4),实验过程中尝试用乙醇钠和叔丁醇钾脱氢没有获得关环产物,必须使用强碱LDA,可能是由于α-H的酸性强度不高的原因. 值得注意的是配体3Lo由于存在分子内盐结构,有一定水溶性,其中和析出过程中要小心控制pH值,否则收率不高.
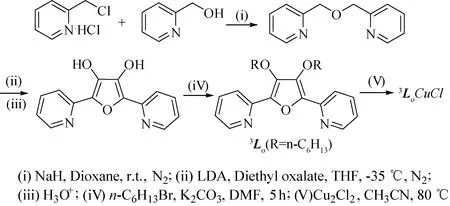
图4 配合物3LoCuCl的合成方案
2.3 配合物3LNCuCl的合成2-吡啶甲硫醚的合成参照文献[13-14],其关环为噻吩衍生物和O-烷基化产物的过程与合成3Lo十分类似(见图5),并且在制备过程中要特别注意避免硫醚的氧化,消除亚砜类杂质的产生.
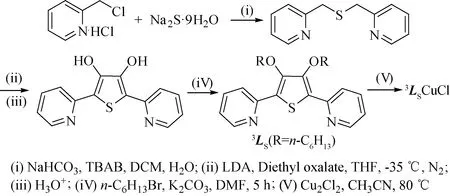
图5 配合物3LSCuCl的合成方案
2.4 甲醇氧化羰基化催化性能比较在120 ℃、总压力2.4 MPa条件下,通过中途取样分别对无催化剂、3LNCuCl、3LoCuCl和3LSCuCl进了连续5 h催化性能监测(包括升温时间30 min),主要产物为DMC,副产物主要是甲缩醛(DMM)和醋酸甲酯(MA).
反应中各种产物的相对含量及增长情况见图6. 无催化剂时没有DMC的生成,而且其DMM和MA含量很低,而且在监测过程中基本维持不变,说明它们在反应初期的升温阶段即产生. 值得注意的是在催化剂3LNCuCl存在时,DMM和MA含量随时间变化并没有明显增加,体现了3LNCuCl极佳的催化选择性. 使用催化剂3LoCuCl和3LSCuCl时,DMM伴随主产物DMC同时生成,MA的产生被抑制. 3种催化剂中3LoCuCl的DMC产生速率明显强于3LNCuCl和3LSCuCl.
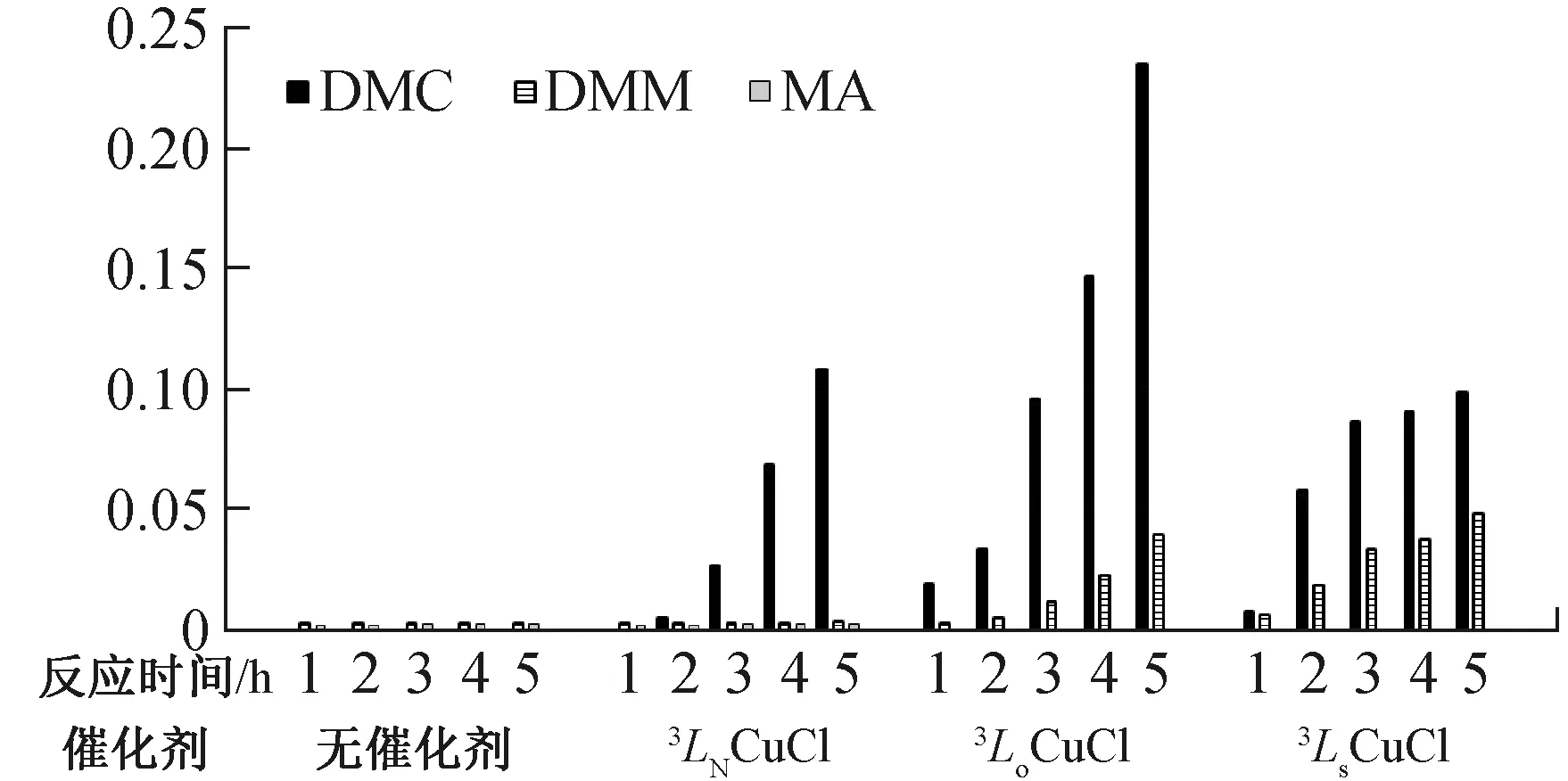
图6 反应产物相对含量
DMC的时空转化率见图7.

图7 DMC时空转化率
反应3 h后,催化剂3LSCuCl对DMC维持约30 g·DMC/(g·cat·h)匀速增加,而3LNCuCl和3LoCuCl对DMC的时空转化速率还在不断增加,5 h后尚未达到最大值,3LoCuCl展示了三者中最佳催化能力. 催化剂活化时间较长的原因可能是螯合物过于稳定,根据配位化合物催化原理,催化过程中存在配位原子与中心离子的脱配位离解的阶段,配位能力过强不利于催化过程中配位键的离解-配位的快速转化. 由于配位能力S-Cu>N-Cu>O-Cu,导致具有N-S-N型三齿配体的3LSCuCl活性不足,而有N-O-N型三齿配体的3LoCuCl在三者中配位键的离解-配位转化速率最快,体现了更好的催化活性.
3种催化剂对DMC的选择性实验结果见图8. 反应3 h后,3LoCuCl的选择性基本保持在85%左右,3LNCuCl增长至90%以上.3LSCuCl的选择性最低,在70%左右,并有降低趋势,可能原因是DMC的生成增长速率过低,而副产物DMM含量增长较快.

图8 DMC选择性
催化剂的选择性与催化剂分子结构特点有关,3LoCuCl和3LSCuCl使用了双吡啶配位,而3LNCuCl则利用了双苯并咪唑配体,3LNCuCl的选择性更好可能是电子云密度较大的咪唑配位的原因,也有可能是空间位阻较大的苯并咪唑更有优势.
3 结论
本研究模拟生物蛋白酶活性中心,设计并合成了3种结构新颖的配合物催化剂3LNCuCl、3LoCuCl和3LSCuCl,用之于甲醇氧化羰基化合成DMC的催化反应研究. 催化剂3LoCuCl显示了对DMC良好的催化合成效果,反应5 h后DMC的时空转化率高于81 g·DMC/(g·cat·h),但是其低于90%的选择性不理想. 催化剂3LNCuCl活化后DMC选择性在94%以上,时空转化率仅为37.5 g·DMC/(g·cat·h),不过催化剂的活化效果在5 h内尚未达到最佳状态,需延长时间进一步确证.在催化剂改进方面,可以尝试结合3LNCuCl和3LoCuCl结构特点的优势,在保证高催化效率的基础上,提高产物选择性,制备综合催化效果更好的工业应用催化剂.
猜你喜欢
分子催化(2022年3期)2022-08-13
渤海大学学报(自然科学版)(2022年1期)2022-07-25
小雪花·初中高分作文(2017年10期)2018-05-15
今日农药(2017年10期)2017-11-14
进出口经理人(2017年12期)2017-10-23
今日财富(2017年32期)2017-10-19
山东工业技术(2017年12期)2017-07-06
科技创新导报(2016年30期)2017-03-15
当代化工(2015年6期)2015-10-21
中学生数理化·高二版(2008年5期)2008-11-12
