
A novel homozygous RAG1 mutation in a girl presenting with granulomas and alopecia capitis totalis
2022-06-30 03:38:48YuRuanQinZhaoQingLiuHongYiZhaoZhiYongZhangYuanDingXiaoDongZhao
World Journal of Pediatrics 2022年4期
Yu Ruan 1,2 · Qin Zhao 2 · Qing Liu 2 · Hong-Yi Zhao 2 · Zhi-Yong Zhang · Yuan Ding 1,2 · Xiao-Dong Zhao
Recombination-activating gene 1 (RAG1
) mutations in humans vary in residual recombination activity and result in heterogeneous clinical phenotypes [ 1, 2]. In recent years,RAG
deficiency with a milder clinical course and delayed presentation has been reported. These patients present with generalized granulomas, severe complications after viral infections, hypogammaglobulinemia, and various autoimmune manifestations (such as cytopenias, vitiligo, psoriasis,myasthenia gravis, and Guillain-Barré syndrome) [ 3- 17].Herein, we analyze the clinical and immunological characteristics of a late-onset combined immunodeficiency caused byRAG1
mutation.The described patient had been vaccinated as planned(including measles, rubella, mumps and varicella zoster virus). She had recurrent thrush in her first 2 years. Then, she suffered from severe chickenpox at 3 years of age and severe adenovirus pneumonia at 5 years of age. At 8 years of age,she got mumps and presented with hair loss. Approximately 6 months later, she was diagnosed with alopecia capitis totalis(Fig. 1 a) and treated with prednisone, but the condition was not improved. During the following 2 years, she was admitted to hospital because of right inguinal lymph node progressive enlargement, right lower limb swelling, and cough. Biopsy of lymph nodes showed lymph node tuberculosis (Fig. 1 c,d). As computed tomography showed bilateral pulmonary lesions (Fig. 2 a-c), mediastinal lymph node enlargement, and multiple low-density areas in the liver and spleen (Fig. 2 d),she was diagnosed with pulmonary tuberculosis and lymph node tuberculosis, despite purified protein derivative test,interferon-gamma release assay and examination of mycobacterium tuberculosis in sputum were negative. Then, she received standard antituberculosis therapy for 10 months, the pulmonary manifestations had no improvement. During this period, she presented with numerous new dark red papules,nodules, and ulcers on the right lower extremity skin, and skin biopsy finally showed cutaneous tuberculosis (Fig. 1 b,e, f). At 10 years of age patient presented severe headaches and convulsions without fever, and cranial magnetic resonance imaging revealed multiple intracranial lesions (Supplementary Fig. 1). Because the treatment was not effective,doctors began to consider whether there was a problem with the patient’s immune function.
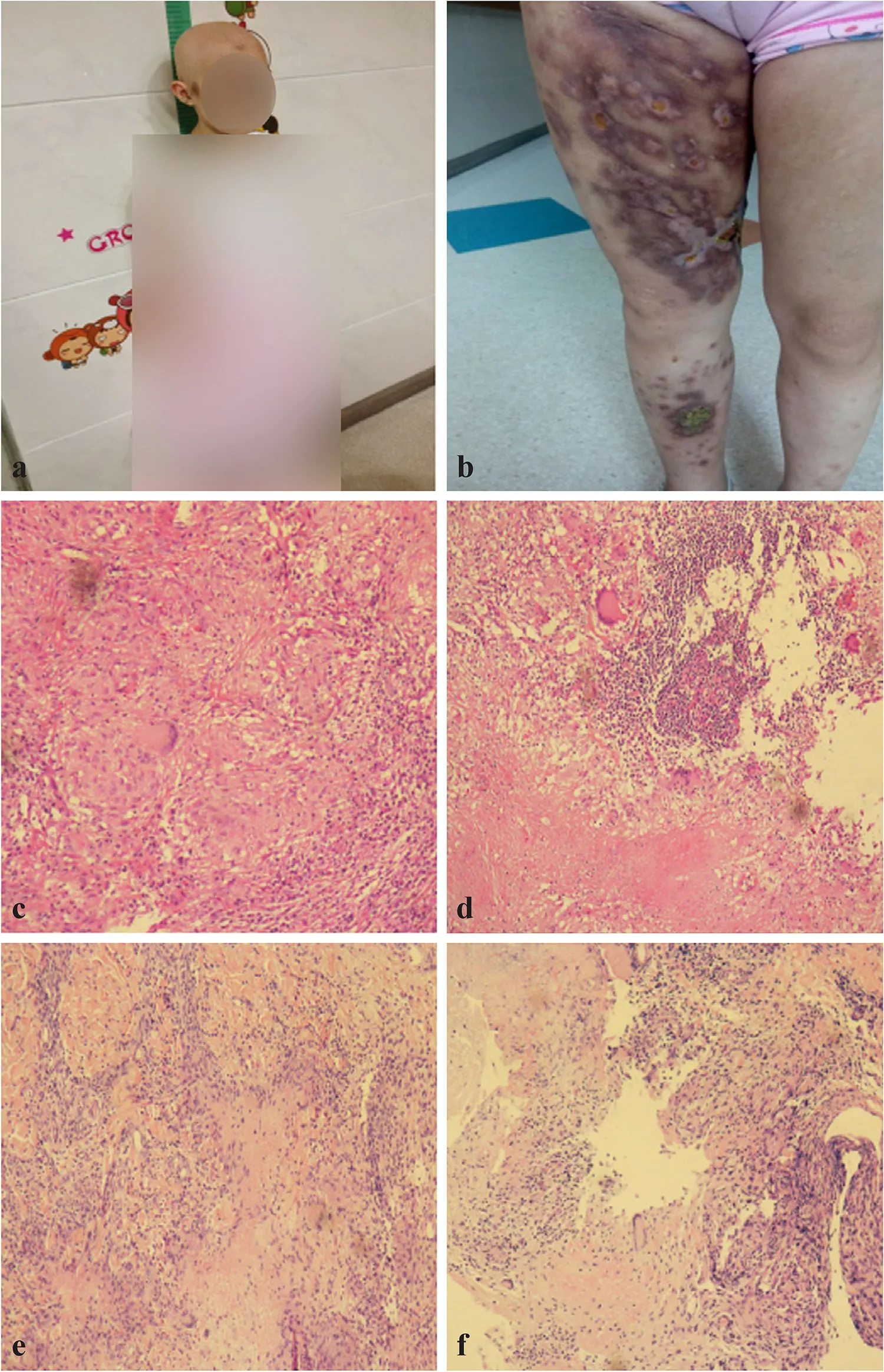
Fig. 1 Alopecia capitis totalis, right lower extremity lesions, and histopathological specimens of the patient. a Alopecia capitis totalis when the patient was 8 years 6 months; b dark red nodules and ulceration on the right lower limb when the patient was 9 years 9 months; c, d lymph node biopsy demonstrated multiple epithelioid granulomas with Langerhans giant cells and a central zone of necrosis; e, f skin biopsy demonstrated granulomatous inflammation involving necrosis and Langerhans giant cells, surrounded by many lymphocytes. Acid-fast stains for mycobacteria were negative (informed consents for publication were obtained from the patient’s legal guardian)

Fig. 2 Computed tomography (CT) images of the patient. Chest CT imaging showed multiple bilateral pulmonary lesions ( a-c); abdominal CT imaging showed multiple low-density areas in the liver ( d)
Phenotyping of patient’s peripheral blood lymphocytes revealed T cell lymphopenia, B cell lymphopenia, and increased levels of natural killer cells.Detailed lymphocyte subsets showed naïve CD4T cells (CD3 + CD4 + CD45RA + CD27 + ), naïve CD8 + T cells(CD3 + CD8 + CD45RA + CD27 + ), central memory CD8 + T cells (CD8 + CD45RA - CD27 + ), and all subsets of B cells were decreased. However, serum levels of immunoglobulin (Ig)G, IgA, IgM, and IgE were normal (Table 1). Serologic antinuclear antibodies, anti-double-stranded DNA antibodies, and antineutrophil cytoplasmic autoantibodies were negative. In this patient, signal joint T-cell receptor (TCR) rearrangement excision circles (per 1 × 10 5 CD3 + T cells) were undetectable compared to those of age-matched healthy controls (253.5 ± 21.95), meaning that V(D)J rearrangement and thymic output were strikingly impaired. Complementarity determining region 3 spectratyping analysis of TCR Vβ was done in total peripheral T cells from this patient and seven healthy control subjects. As shown in Supplementary Fig. 2, seven Vβ subfamilies from this patient displayed monoclonal or oligoclonal TCR Vβ repertoires, indicating skewed spectratyping profiles, while the majority of Vβ subfamilies from healthy controls indicated polyclonal TCR Vβ repertoires. Phytohemagglutinin-induced proliferation of CD4 +and CD8 + T cells in this girl was undetectable compared with her mother (Supplementary Fig. 3). Taken together,these results indicate that the development and function of T cells were impaired in this patient. Finally, genetic analysis of this patient demonstrated a novel homozygous missense mutation (c.2971 C > G, p. L991V) in exon 2 of theRAG1
gene (NM_00048, Fig. 3). The patient did not receive regular treatments and follow-up for economic reasons. She died at the age of 11 years from respiratory and cardiac arrest caused by sputum blocking.Granulomatous lesions have been reported in several patients with hypomorphicRAG1
mutations. The localization of granuloma varied from skin to internal organs (e.g.,lungs, tongue, adenoids, spleen, liver, colon) and age of granuloma formation from 2 to 14 years [ 1, 4- 8, 13- 15].In described patients granulomas are mostly necrotizing[ 4, 5, 7, 13], which needs to be differentiated from tuberculous granulomas and other granulomatous diseases (e.g.,chronic granulomatous disease, Blau syndrome, Wegener's granulomatosis, and Crohn’s disease). In our case,granulomas were identified in the skin and lymph node,and the age of granuloma formation was 9 years. There were also multiple lesions in the lung, brain, liver, and spleen, and it is not clear whether they were caused bygranulomatous inflammation, infection, or infection combined with granuloma.RAG1
mutations associated with combined immunodeficiency associated with granulomas and/or autoimmunity were mostly missense mutations which were predominantly located in the dimerization and DNA-binding domain, pre-RNase H and carboxy-terminal domain (CTD) [ 3]. Three mutations (p. F974L, p. R975W and p. R975Q) in the CTD of RAG1 protein have been reported [ 4, 17, 18]. In this case, the mutation (p. L991V),which was located in the CTD of the protein, has not been reported previously. Studies on the genotype-phenotype correlation in humanRAG1
deficiency are ongoing. It is suggested that the clinical phenotype is determined by howRAG
mutations affect immune system development and function and by immune responses to environmental factors [ 3, 16].Table 1 Hematologic and immunologic characteristics of the patient
T cell receptor, immunoglobulin, double negative T cell, quantification of signal joint TCR rearrangement excision circle a Listed are reference ranges or laboratory values for the patient’s age group. “-” no data
Parameters 8 years 11 months 10 years 1 months 10 years 10 months Reference range a White cell count Leukocytes (cells × 10 3 /μL) 6.47 5.05 14.98 4.00-10.00 Lymphocytes (% of leukocytes) 17 13 6 30-60 Neutrophils (% of leukocytes) 72 83 90 33-79 Eosinophils (% of leukocytes) 4 2 1 0-5 Lymphocyte subsets, % (cells/μL)CD3 + 39.0 (429.0) 24.0 (157.6) 31.3 (281.5) 54.6-79.3 (1131.1-2766.9)CD3 + CD4 + 22.0 (242.0) 11.2 (73.5) 17.0 (153.4) 22.0-42.5 (497.1-1404.5)CD3 + CD4 + CD45RA + CD27 + - - 1.7 (15.3) 6.1-27.3 (156.7-791.5)CD3 + CD4 + CD45RA + CD27 - - - 0.6 (5.1) 0-1.6 (0-43.9)CD3 + CD4 + CD45RA - CD27 + - - 12.2 (109.8) 5.6-21.0 (160.3-592.8)CD3 + CD4 + CD45RA - CD27 - - - 2.6 (23.0) 0.8-5.0 (19.4-129.1)CD3 + CD8 + 14.0 (154.0) 8.3 (54.5) 12.0 (107.6) 18.5-36.9 (413.8-1323.6)CD3 + CD8 + CD45RA + CD27 + - - 5.6 (50.7) 6.2-21.7 (186.6-738.9)CD3 + CD8 + CD45RA + CD27 - - - 4.4 (39.4) 0.1-8.2 (2.4-214.0)CD8 + CD45RA - CD27 + - - 1.7 (15.1) 3.0-13.4 (64.5-403.1)CD3 + CD8 + CD45RA - CD27 - - - 0.3 (2.4) 0.2-9.6 (5.7-321.7)TCRαβ + DNT - - 0.1 (0.7) 0.5-2.8 (11.2-71.6)CD3 + TCRγδ + - - 4.4 (40.0) 3.8-22.1 (97.0-631.3)CD19 + 3.0 (33.0) 3.1 (20.4) 1.0 (8.9) 6.9-20.5 (154.1-624.7)CD19 + CD27 + - - 0.2 (1.9) 0.9-4.4 (16.5-123.7)CD19 + CD27 - IgD + - - 0.0 (0.1) 3.9-14.9 (102.3-452.2)CD19 + CD24 ++ CD38 ++ - - 0.0 (0.0) 0.1-1.8 (2.8-45.6)CD19 + CD24 - CD38 ++ - - 0.0 (0.0) 0.1-2.4 (3.1-51.6)CD16 + CD56 + 54.0 (594.0) 72.0 (472.7) 65.9 (592.7) 8.0-31.8 (195.0-1150.4)Immunoglobulins IgG (g/L) 13.30 18.50 - 5.28-21.9 IgA (g/L) 1.48 1.45 - 0.51-2.59 IgM (g/L) 1.36 1.23 - 0.48-2.26 IgE (IU/mL) 1.50 0.30 - < 150.00 sjTRECs (copies per 1 × 10 5 CD3 + T cells) - - Undetectable T cell proliferation - - Undetectable T cell receptor repertoires - - Restricted Vβ repertoire

Fig. 3 Pedigree and mutation analysis of a recombination-activating gene 1-deficient patient
In summary, we report a late-onset combined immunodeficiency phenotype characterized by granulomatous inflammation and alopecia capitis totalis, with a novel homozygous missense mutation inRAG1
. OnlyRAG
mutations with severe combined immunodeficiency disorders(SCID) or Omenn syndrome have been reported in the Chinese population; thus, this case broadens the spectrum of disorders associated withRAG1
mutations and may have important implications for early diagnosis and treatment.Besides, this condition is expected to be recognized early by the newborn screening for SCID. Moreover, clinicians should recognize the clinical warning signs of primary immunodeficiency disease (such as recurrent infections,granulomatous inflammation, autoimmunity, persistent lymphopenia, and poor therapeutic effect, etc.), include immune deficiency in the differential diagnosis and conduct diagnosis confirmation by genetic analysis when necessary.Supplementary Information
The online version contains supplementary material available at https:// doi. org/ 10. 1007/ s12519- 021- 00503-3.Acknowledgements
We are grateful to the patient and family for their kind cooperation in this study.Author contributions
RY conceptualized and designed the study, and performed data analysis and drafted the initial manuscript. ZQ, LQ and ZHY helped design the study, collected data and assisted with data analysis, and critically reviewed and revised the manuscript. ZZY and DY conceptualized and designed the study, assisted with data interpretation and supervised the study, and critically reviewed and revised the manuscript. ZXD conceptualized and designed the study, and critically reviewed and revised the manuscript. All authors approved the final version of the manuscript.Funding
This study was supported by the Public Welfare Scientific Research Project of China (No. 201402012) and the National Natural Science Foundation of China (No. 81974255).Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.Declarations
Ethical approval
This research was approved by the Ethics Committee of the Children’s Hospital of Chongqing Medical University (approval number: 001/2013).Conflict of interest
No financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article. Author Xiao-Dong Zhao is a member of the Editorial Board forWorld Journal of Pediatrics.
The paper was handled by the other Editor and has undergone rigorous peer review process. Author Xiao-Dong Zhao was not involved in the journal's review of, or decisions related to, this manuscript.Patient consent statement
Informed consent was obtained from the patient’s legal guardian. World Journal of Pediatrics2022年4期
World Journal of Pediatrics2022年4期
- World Journal of Pediatrics的其它文章
- Role of Omicron variant of SARS-CoV-2 in children in Germany
- Continuing interventions in a quality improvement bundle to reduce bronchopulmonary dysplasia
- Catheter-related bloodstream infections in children with intestinal failure: a 6-year review from an intestinal rehabilitation center in China
- Different approaches for patent ductus arteriosus in premature infants using acetaminophen
- Therapeutic benefits of zinc sulfate on neonatal hyperbilirubinemia
- ALK expression, prognostic significance, and its association with MYCNexpression in MYCN non-amplified neuroblastoma