
硅钛杂化介孔球负载金纳米粒子及其催化性能调控
2022-06-29 08:47陶疆辉王艳妮王亚斌丁秀萍
无机材料学报 2022年4期
马 慧,陶疆辉,王艳妮,韩 玉,王亚斌,2,丁秀萍
(1.延安大学 化学与化工学院,陕西省化学反应工程重点实验室,延安 716000;2.西北工业大学 化学与化工学院,西安 710129;3.中国科学院 青海盐湖研究所,中国科学院盐湖资源综合高效利用重点实验室,西宁 810008)
近些年,负载型贵金属催化剂在设计、制备、应用及理论研究方面取得了长足的进步[1]。通过调控以下几个因素进行催化剂设计: (1)载体的化学组成。迄今为止,多种材料被开发用作载体平台,如传统的二氧化硅(SiO2)[2]、二氧化钛(TiO2)[3]、碳[4],以及新型材料金属有机框架(MOF)[5]、碳氮化合物(C3N4)[6]等。值得注意的是,TiO2和C3N4本身就具备催化性能。(2)载体中元素的化学态。Wang 等[7]制备了氮掺杂的碳载体,并且发现石墨相的氮能够促使贵金属成核及分散,而吡啶氮则帮助分散剂锚定贵金属。(3)载体的形貌及结构。Peng 等[8]制备了系列二氧化硅基载体负载银纳米粒子用于催化氧化一氧化碳,包括传统六方有序结构的MCM-41 和SBA-15,以及新型树枝状介孔二氧化硅纳米球(Dendritic mesoporous silica nanospheres,DMSNs)。研究表明载体结构及形貌可以控制银纳米粒子的晶粒尺寸和分散性,进而影响催化活性。(4)载体表面功能性。通过化学改性,载体界面可以被赋予与贵金属纳米粒子相同优异亲和性的理想有机官能团[9],官能团与贵金属的化学作用直接影响其物理化学状态。以上因素独立或共同控制贵金属纳米粒子的负载量及分散程度,进而影响催化性能。
树枝状介孔二氧化硅纳米球拥有独特的三维中心辐射状孔道及多级孔结构,具有大的比表面积和孔体积[8,10-11],使其广泛用于负载纳米尺寸的客体物质,用作新型的载体/递送/反应平台[12-14]。研究表明: 相对于传统MCM-41 和SBA-15,DMSNs 能够负载更多的贵金属纳米粒子[8,10-11]。众多研究小组通过化学改性在DMSNs 表面嫁接有机官能团,然后通过浸渍法和硼氢化钠还原技术,将贵金属纳米粒子锚定在放射状孔道,兰州大学董正平教授团队在此领域开展了很多基础研究[9,15-17]。
本工作以DMSNs 的特异形貌及结构为基础,通过调控其化学组成及表面功能性,构筑新型负载型贵金属催化剂。以纯二氧化硅组成的DMSNs 为基础,引入TiO2化学组成,形成树枝状介孔硅钛杂化纳米球(Dendritic mesoporous silica&titania nanospheres,DMSTNs)。通过有机改性方法在其表面接枝3-氨基丙基三乙氧基硅烷(DMSTNs-NH2),氨基功能化的载体浸渍吸附金离子前驱体并采用硼氢化钠还原方法,得到在DMSTNs 放射孔道负载金纳米粒子的复合纳米催化剂(DMSTNs-NH2-Au)。以DMSNs-NH2-Au 为对照样品,考察两类负载型催化剂的基本物理化学性能、结构形貌特征,模拟太阳光下催化裂解水产氢性能及无光条件下对硝基苯酚的还原性能。
1 实验方法
1.1 材料制备
DMSNs 及DMSTNs 的制备可参照本课题组前期研究工作[18-20],其表面接枝氨基官能团可采用传统有机化学改性方法[15,21]。以DMSTNs-NH2为例,将0.5 g 粉末状DMSNs 或DMSTNs 超声分散于50 mL 无水甲苯溶液,逐滴加入2.5 mmol 的3-氨基丙基三乙氧基硅烷(分析纯,99%),在氮气保护条件下加热至105 ℃回流12 h。离心分离产物用无水乙醇清洗4 次,在真空干燥箱30 ℃烘干备用。
负载金纳米粒子的实验流程如下: 取1.0 g 氨基功能化DMSNs-NH2或DMSTNs-NH2,超声分散于100 mL 去离子水。将含0.5 mmol 金离子的氯金酸溶液(10 g/L)逐滴加入上述悬浊液并搅拌45 min。再逐滴加入60 mL 浓度为100 mmol/L 的新制NaBH4溶液,混合液变为褐色。充分搅拌反应4 h,产物离心分离,用去离子水洗涤4 次,在真空干燥箱35 ℃干燥10 h。制得的目标产物分别记为DMSNs-NH2-Au和DMSTNs- NH2-Au (图1)。

图1 DMSNs (a)与DMSTNs (b)负载金纳米粒子催化剂的制备流程示意图Fig.1 Schematic diagram of constructing DMSNs- (a) and DMSTN- (b) based supported Au NPs catalysts
1.2 材料表征
采用扫描电子显微镜(SEM,HITACHI-SU8010,Japan)和透射电子显微镜(TEM,JEM-F200,Japan)观察样品的形貌及结构。采用电感耦合等离子体发射光谱仪 (ICP,PerkinElmer Optima 5300 DV,USA)测定金含量。采用X 射线光电子能谱仪(XPS,Thermo Fisher Scientific,USA)分析材料表面元素及其化学态。采用傅里叶转换红外光谱仪(FT-IR,NEXUS-870,USA)检测材料表面有机官能团。采用光致发光光谱(PL,F-4500,Hitachi,Japan)研究催化剂电子结构对催化性能的影响。采用紫外-可见漫反射光谱(UV-Vis-DRS,UV-2550,shimadzu,Japan)研究样品光吸收性能。
1.3 催化性能表征
在多通道光反应器(Perfectlight PCX-50B Discover)中测试样品的光催化裂解水产氢,流程如下: 将60 mg 的固体粉末加入到80 mL 质量分数10%的三乙醇胺水溶液,超声45 min 后放入反应瓶中。将反应瓶置于气氛控制器中,通入氮气,排除瓶内的空气,然后放入多通道光催化反应装置中。光照射5 h(AM1.5 光源),使用气相色谱仪在线监测产生的气体。在紫外可见分光光度计(UV-2550,shimadzu,Japan)中还原对硝基苯酚,流程如下: 将新制的2.5 mL 浓度为0.12 mmol/L 对硝基苯酚溶液加入比色皿,快速加入新制的500 μL 0.5 mol/L 硼氢化钠溶液,再将10 μL 0.5 mg/mL 催化剂悬浊液快速加入上述混合液,在不同时间点记录吸收曲线。
2 结果与讨论
图2为 DMSNs、DMSTNs、DMSNs-NH2、DMSTNs-NH2、DMSNs-NH2-Au 和DMSTNs-NH2-Au的SEM 照片。六种纳米材料单分散无团聚,均呈现规则的球状。纳米褶皱结构形成了辐射状孔道,球体表面具有不同尺寸的纳米孔。DMSNs(图2(a))负载二氧化钛,孔道被填充,使DMSTNs 孔径明显减小(图2(b))。经过有机硅烷改性的DMSNs-NH2(图2(c))和DMSTNs-NH2(图2(d)),以及负载金纳米粒子的催化剂DMSNs-NH2-Au(图2(e))和DMSTNs-NH2-Au(图2(f)),在形貌上均无巨大变化,很难观察出明显的差异。

图2 DMSNs (a)、DMSTNs (b)、DMSNs-NH2 (c)、DMSTNs-NH2 (d)、DMSNs-NH2-Au (e)和DMSTNs-NH2-Au (f)的SEM 照片Fig.2 SEM images of DMSNs (a),DMSTNs (b),DMSNs-NH2 (c),DMSTNs-NH2 (d),DMSNs-NH2-Au (e),and DMSTNs-NH2-Au (f)
采用傅里叶转换红外光谱仪检测DMSNs 和DMSTNs 表面官能团组成,由图3可见: DMSNs 在1090、1645、3430 cm-1处出现了Si-O-Si、H2O、-OH(Si-OH 或/和Ti-OH)的吸收峰[22]。DMSTNs 在530~630 cm-1范围内具有Ti-O-Ti 的特征平峰[23]。氨基功能化的DMSNs 和DMSTNs 仍然具有上述特征吸收峰,表明两类材料骨架表面化学性能十分稳定;3430 cm-1处的吸收急剧减弱,表明两种材料与硅烷分子中的硅羟基脱水缩合。DMSNs-NH2和DMSTNs-NH2在1380~1500 cm-1和2930~3000 cm-1处的吸收峰对应-CH2-结构的弯曲振动和伸缩振动,证明硅烷骨架已接枝在样品表面。理论上在3300 cm-1处的-NH2特征峰被-OH 缔合宽峰覆盖,可以使用XPS 来确定表面氨基化。

图3 DMSNs、DMSTNs、DMSNs-NH2 和DMSTNs-NH2 的傅里叶转换红外光谱图Fig.3 FT-IR spectra of DMSNs,DMSTNs,DMSNs-NH2,and DMSTNs-NH2
图4为DMSTNs、DMSNs-NH2和DMSTNs-NH2的XPS 全谱扫描图。杂化样品含有Ti2p 和Ti3p 特征峰,硅烷改性样品C1s 特征峰强度明显增大,在约400 eV 结合能处可以清晰观察到氨基硅烷改性样品表面出现N1s 特征峰。X 射线光电子能谱和红外光谱结果共同确定DMSNs 和DMSTNs 表面成功接枝了氨基官能团,这为载体提供了活性位点。

图4 DMSTNs、DMSNs-NH2 和DMSTNs-NH2 的XPS 全谱扫描Fig.4 XPS survey scans of DMSTNs,DMSNs-NH2,and DMSNs-NH2
对六种纳米材料进行透射电子成像分析,它们的骨架均呈现出中心辐射状纹理,形成不同尺寸大小的孔道。纯二氧化硅基DMSNs(图5(a))生成的氨基功能化DMSNs-NH2(图5(c))结构纹理不变。金纳米粒子负载于孔道后,可以清楚观察到纳米球孔道负载了纳米粒子(图5(e2))。高分辨透射电镜(图5(e3))检测出金纳米粒子的特征晶格条纹。结合透射电镜-元素分布图(图5(g)),可以充分证明金纳米粒子均匀分散在球体的辐射状孔道内。DMSTNs(图5(b))和DMSTNs-NH2(图5(d))孔道明显填充了尺寸均匀的纳米粒子,本课题组前期工作已证明其为直径约9.0 nm 的TiO2纳米颗粒[18]。金纳米粒子是否锚定在氨基功能化硅钛杂化球孔道内,仅通过透射电镜分析难以确定(图(5f1,f2))。通过高分辨透射电镜(图5(f3))证明存在TiO2和Au 纳米粒子。元素分布图(图5(h))可以看出金纳米粒子也均匀分散在辐射状孔道内。DMSTNs-NH2-Au 在XRD 图谱(图6(a))中也出现了锐钛矿TiO2的特征峰(101)、(200)和(211),以及金纳米粒子的特征峰(111)、(200)、(220)和(311)。以上结果表明:(1)DMSNs和DMSTNs化学骨架稳定性强,能够耐受有机溶液甲苯改性和无机硼氢化钠水溶液还原环境;(2)氨基能够成功嫁接且均匀分布在 DMSNs 和DMSTNs 表面;(3)金纳米粒子被氨基成功锚定且均匀分散于功能化DMSNs和DMSTNs的辐射状孔道内。

图5 DMSNs (a)、DMSTNs (b)、DMSNs-NH2 (c)、DMSTNs-NH2 (d)、DMSNs-NH2-Au (e)、DMSTNs-NH2-Au (f)的透射电镜照片,DMSNs-NH2-Au (g)和DMSTNs-NH2-Au (h)的透射电镜-元素分布图Fig.5 TEM images of DMSNs (a),DMSTNs (b),DMSNs-NH2 (c),DMSTNs-NH2 (d),DMSNs-NH2-Au (e),and DMSTNs-NH2-Au (f),and TEM-mapping images of DMSNs-NH2-Au (g) and DMSTNs-NH2-Au (h)
电感耦合等离子体发射光谱仪测得DMSNs-NH2-Au 和DMSTNs-NH2-Au 中金的质量分数分别为6.96%与6.66%,两者相差不大。图6(b)为DMSNs、DMSTNs、DMSNs-NH2-Au 和DMSTNs-NH2-Au 的紫外可见漫反射图谱。DMSNs 在200~800 nm 整个波段没有吸收,近似为一水平线。DMSTNs 在200~400 nm 紫外区域表现出宽吸收,可归因于Si-O-Ti 杂化中的钛四面体结构和Ti-O-Ti 结构中的钛八面体结构[11]。DMSNs-NH2-Au 吸收增强,金纳米粒子的特征吸收也出现在350~600 nm 区域[24]。DMSTNs-NH2-Au 在紫外和可见光区域同时具有DMSTNs 和Au 纳米粒子的特征吸收。
图6(c)为DMSNs、DMSTNs、DMSNs-NH2-Au和DMSTNs-NH2-Au 的光致发光图谱。绝缘性DMSNs在300~500 nm 波段光致发光谱表现为一条逐渐下降的曲线。硅钛杂化DMSTNs 在325~500 nm 波段具有一个宽的吸收峰,DMSNs-NH2-Au 吸收强度较DMSNs 急剧减弱,也趋于一条直线。DMSTNs-NH2-Au 在整个波段的吸收最弱,特征吸收峰几乎消耗殆尽。以上结果表明DMSTNs-NH2-Au 的光生电子-空穴对的复合速率急剧下降,也就是说它们的分离速率增强,这对催化反应十分有利[25]。

图6 DMSNs、DMSTNs、DMSNs-NH2-Au 和DMSTNs-NH2-Au 的XRD 图谱(a),紫外-可见漫反射光谱图(b)和光致发光谱图(c)Fig.6 XRD patterns (a),UV-Vis-DRS spectra (b) and PL spectra (c) of DMSNs,DMSTNs,DMSNs-NH2-Au,and DMSTNs-NH2-Au
图7(a,b)为DMSNs、DMSTNs、DMSNs-NH2-Au和DMSTNs-NH2-Au 在模拟太阳光下裂解水的氢气产量及相应速率。DMSNs 在5 h 内的氢产量和速率分别为50.07 μmol·g-1和10.01 μmol·g-1·h-1。DMSNs中存在的大量碱性活性位点和氧空位使其具有一定催化性能[26]。DMSTNs 的光催化性能降低,氢产量及速率分别为38.06 μmol·g-1和7.61 μmol·g-1·h-1。DMSTNs 催化活性降低的原因如下: (1) 大量碱性活性位点和氧空位被负载的TiO2占据;(2) TiO2的含量有限及光生电子-空穴对快速复合(图6(c));(3)孔体积锐减(图2)。DMSNs-NH2-Au 催化性能最低,氢产量及速率仅为10.06 μmol·g-1和2.01 μmol·g-1·h-1,这是由于表面改性使得氧空位和碱性活性位点被有机单体覆盖,并且负载在孔道内的贵金属Au 纳米粒子不能有效发挥光催化活性。DMSTNs-NH2-Au氢产量及速率为69.08 μmol·g-1和13.82 μmol·g-1·h-1,较DMSNs 和DMSNs-NH2-Au 分别提升1.4 和6.8 倍。经过五次循环产氢,DMSTNs-NH2-Au 的催化性能略微下降(图7(c)),催化剂中心辐射状骨架结构没有遭受破坏(图7(d)),金纳米粒子负载于中心发射状孔道内(图7(d)插图)。图8(a)为对硝基苯酚、对硝基苯酚钠(对硝基苯酚中加入硼氢化钠)和对氨基苯酚(充分还原对硝基苯酚)的紫外可见漫反射光谱,吸收特征峰分别出现在约317、400 和300 nm 位置。图8(b~d)分别为加入硼氢化钠的对硝基苯酚空白溶液、空白溶液加入 DMSNs 以及空白溶液加入DMSTNs 样品在840 s 内的紫外吸收变化。三个体系的吸收峰强度基本无变化,表明在没有催化剂的情况下均很难还原对硝基苯酚。

图7 DMSNs、DMSTNs、DMSNs-NH2-Au 和DMSTNs-NH2-Au 在模拟太阳光下的氢产量(a)及速率(b);DMSTNs-NH2-Au的光解水产氢循环性能(c);DMSTNs-NH2-Au 经过5 次催化循环后的透射电镜照片(插图为高角环形暗场成像)(d)Fig.7 H2 production amount as a function of irradiation time (a) and the corresponding production rates (b) of DMSNs,DMSTNs,DMSNs-NH2-Au,and DMSTNs-NH2-Au,cycling tests of DMSTNs-NH2-Au for H2 production (c),and TEM image of DMSTNs-NH2-Au sample experienced five cycles,with inset showing the high-angle annular dark-field imaging (d)
DMSNs-NH2-Au 能够相对较快地还原对硝基苯酚(图8(e)),而DMSTNs-NH2-Au 在相同时间内表现出优异的还原性能与合成对氨基苯酚的活性(图8(f))。转化率Ct/C0可由400 nm 处的吸光度比值At/A0确定(图8(g)),其中,C0为对硝基苯酚初始浓度,Ct为其在t时刻的浓度。鉴于NaBH4溶液浓度远大于对硝基苯酚浓度(参见实验部分),将NaBH4浓度视为常数,对硝基苯酚还原可依照准一级反应近似处理[16]。由图8(h)可知,-ln(Ct/C0)和反应时间呈直线状,可用-ln(Ct/C0)=kt表示,k为表观动力学常数(s-1)。DMSTNs-NH2-Au 的转化率远优于DMSNs-NH2-Au,表观动力学常数约为后者的17倍。循环利用DMSTNs-NH2-Au 降解对硝基苯酚10 次,延长降解时间为30 min(充分反应),转化率由99.8%下降至98.6%(图8(i),Δ=1.2%)。10 次循环后样品的形貌基本无变化(图8(j)),表明DMSTNs-NH2-Au 在该反应体系具有优越的稳定性。EDS-mapping 证明元素硅、钛、硫和金依旧均匀存在于10 次催化循环的DMSTNs-NH2-Au中(图8(k))。对任意4 个单体球进行元素分析(图8(k1)中l1~l4),它们的构成比例十分接近(EDS 数值)。以上结果充分证明DMSTNs-NH2-Au 样品中元素均匀分散且催化性能十分稳定。
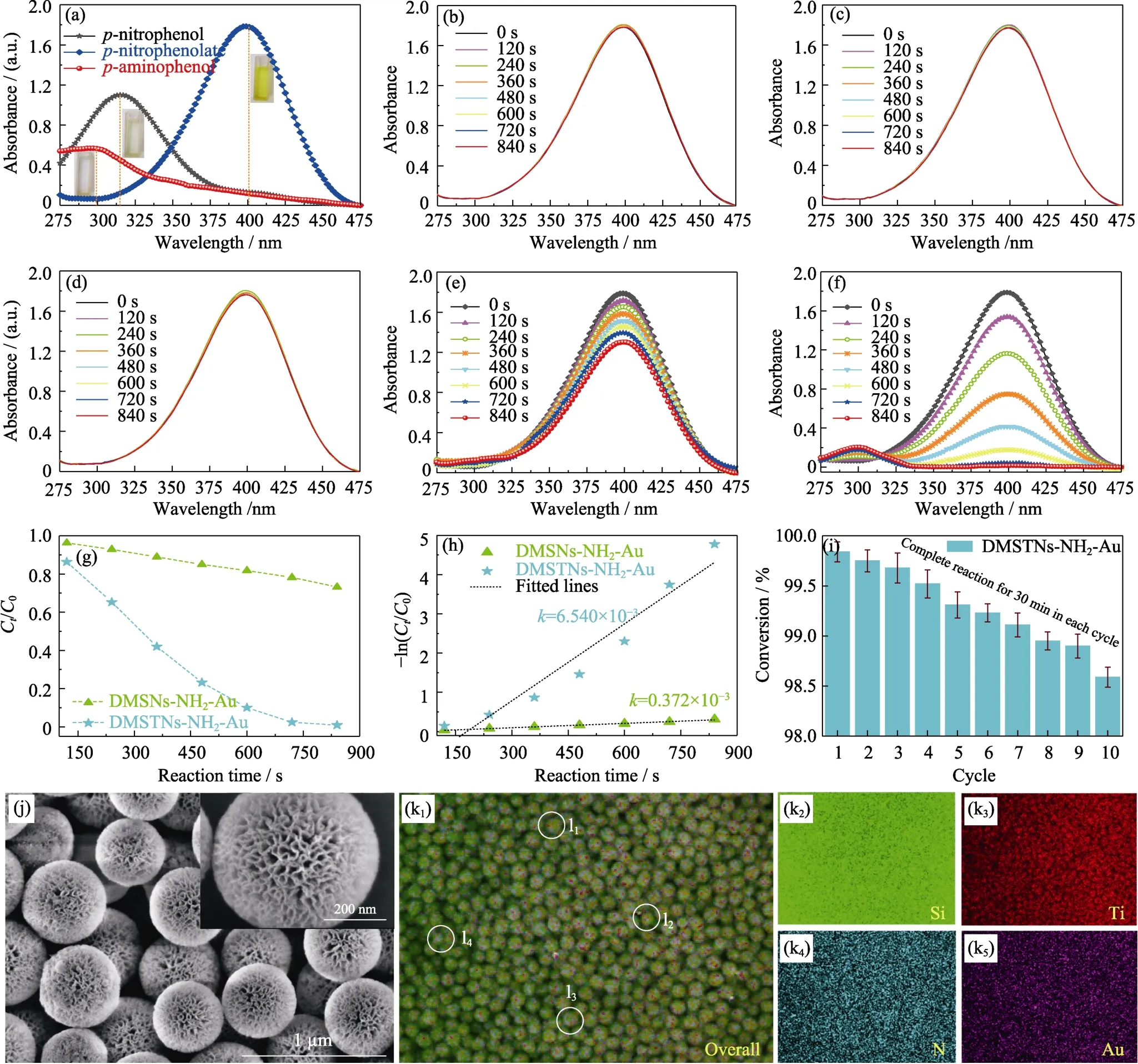
图8 对硝基苯酚、对硝基苯酚钠和对氨基苯酚的紫外可见漫反射光谱(a),不同样品(含有硼氢化钠的对硝基苯酚溶液作为空白试样(b)、加入DMSNs(c)、DMSTNs(d)、DMSNs-NH2-Au(e)和DMSTNs-NH2-Au(f))的紫外吸收变化,DMSNs-NH2-Au和DMSTNs-NH2-Au 的转化率(g)及准一级模拟方程(h),DMSTNs-NH2-Au 循环降解性能(i),DMSTNs-NH2-Au 经过10 次催化循环后的SEM 形貌(j)及元素分布图(k),任意4 个经过10 次催化循环的DMSTNs-NH2-Au 单体球能谱测试标定区域(l)Fig.8 Characteristic ultraviolet absorption peaks of p-nitrophenol,p-nitrophenolate,and p-aminophenol (a),Ultraviolet absorption spectra of different samples,including p-nitrophenol+NaBH4 as the blank sample (b),with the addition of DMSNs (c),DMSTNs (d),DMSNs-NH2-Au (e),and DMSTNs-NH2-Au (f),the conversion (g) and pseudo first-order linear equation (h)of DMSNs-NH2-Au,and DMSTNs-NH2-Au,and cycling tests of DMSTNs-NH2-Au for p-nitrophenol reduction (i),SEM images (j) and energy dispersive spectroscopy (EDS) mappings (k) of DMSTNs-NH2-Au sample experienced ten cycles.EDS measurement of random four DMSTNs-NH2-Au individuals (l)
图9为DMSTNs-NH2-Au 在模拟太阳光下产氢及无光条件下还原对硝基苯酚的催化机理。在模拟太阳光照射下,DMSTNs-NH2-Au 的催化机制分为两种情况(图9(a)): 在紫外光区域,高能光子可激发光生电子从二氧化钛的价带转移至导带,伴随产生的价带电子空穴能够提供正电荷使水生成氢离子。导带电子迁移至金纳米粒子的表观费米能级(EF*)[27-28],与氢离子结合还原为H2;在可见光区域,低能光子没有足够能量激发电子从二氧化钛的价带转移至导带。但是,金纳米粒子吸收光子后产生了局域表面等离子体共振效应(Localized Surface Plasmon Resonance,LSPR)[29-31],电子可从其表观费米能级转移至二氧化钛的导带。金纳米粒子正电位促使水产生氢离子,二氧化钛导带提供电子使得氢离子变为氢气。在无光照条件下,BH4-提供的大量电子可通过金纳米粒子的表观费米能级转移至二氧化钛导带[32-33],对硝基苯酚接受电子被还原为对氨基苯酚(图9(b))。总之,光生电子和来自BH4-的电子经历了更长的迁移路程,这些变化大幅提升催化活性。

图9 DMSTNs-NH2-Au 在(a)模拟太阳光条件下光解水制氢和(b)无光照条件下还原对硝基苯酚的催化机理Fig.9 Schematic illustration of possible photocatalytic mechanisms for DMSTNs-NH2-Au to split water under simulated sunlight (a) and ordinary catalytic reduction of p-nitrophenol without light irritation (b)
3 结论
本工作成功构筑了一类结构稳定的树枝状硅钛杂化纳米球负载金纳米颗粒多功能复合催化剂材料。树枝状中心辐射孔道内成功负载了均匀分散的锐钛矿二氧化钛纳米粒子和超小金纳米颗粒。相较于树枝状二氧化硅纳米球负载金纳米颗粒对比样催化剂材料,新型催化剂展现出更加优越的多功能催化性能。在模拟太阳光下,其光解水产氢量及速率为69.08 μmol·g-1和13.82 μmol·g-1·h-1,约为对比样品的7 倍。在无光条件下,其降解对硝基苯酚的表观动力学常数为6.540×10-3s-1,约为对比样品的17 倍(0.372×10-3s-1)。中心放射树枝状纹理结构、增加的光生电子-空穴对分离速率以及更长的电子迁移路程大幅提升了DMSTNs-SH-Au 的催化活性。
猜你喜欢
云南化工(2022年9期)2022-10-12
农药学学报(2022年3期)2022-06-14
节能与环保(2022年3期)2022-04-26
安徽农学通报(2022年6期)2022-04-07
内燃机与动力装置(2022年1期)2022-03-21
中国新技术新产品(2020年5期)2020-05-06
农业工程学报(2020年3期)2020-04-09
现代农业科技(2018年23期)2018-02-18
筑路机械与施工机械化(2017年5期)2017-08-31
家庭用药(2016年8期)2016-05-14
