
QuEChERS净化-柱前衍生-高效液相色谱法测定烟叶中抗蚜威的残留量
2022-06-24 02:01王焕琦聂四平黄家岭王正强
理化检验-化学分册 2022年6期
王焕琦 ,聂四平 ,黄家岭 ,王正强
(1.贵州省产品质量检验检测院,贵阳 550016;2.贵州医科大学,贵阳 550025)
抗蚜威是以甲酸酯为前体化合物合成得到的一种农药[1],结构见图1。
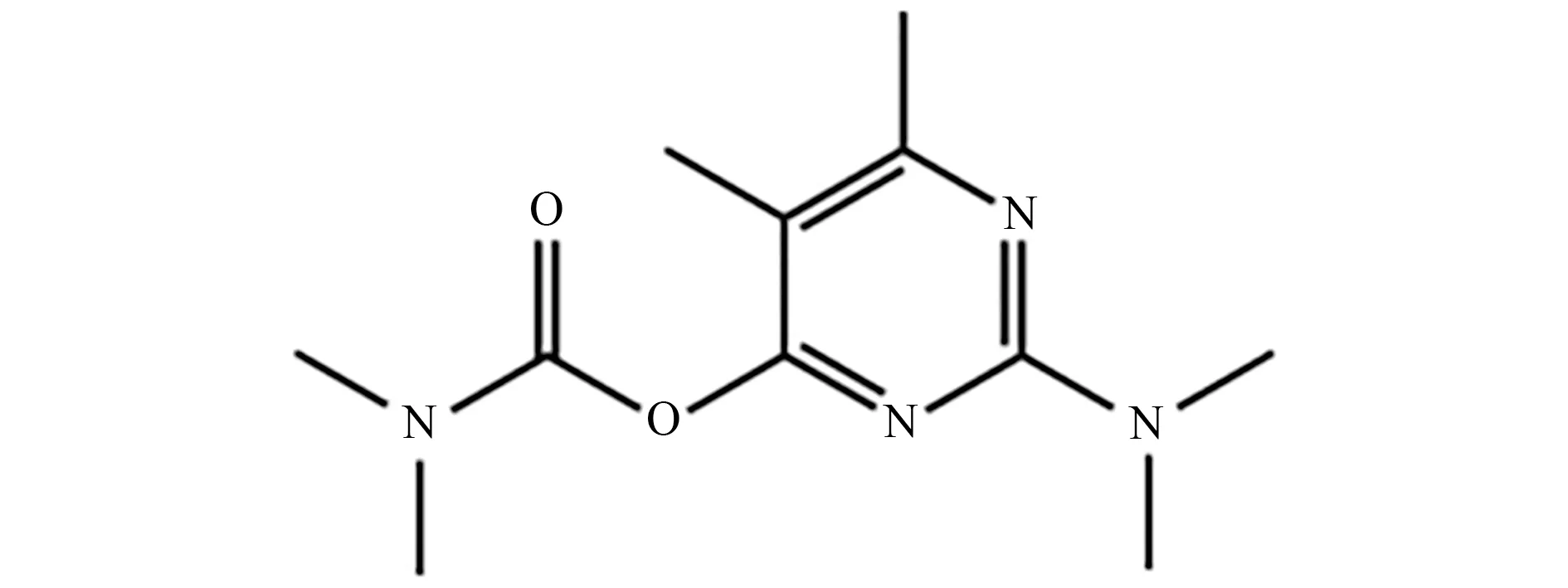
图1 抗蚜威的结构Fig.1 Structure of pirimicarb
灭虫原理是抑制乙酰胆碱酯酶在昆虫体内的活性,从而降低昆虫的神经传导功能。它是一种神经内毒素,有一定的遗传毒性和细胞毒性[2-3],广泛应用于农业生产,具有灭虫效果明显、选择性高和残留期短等特点,但是若大量使用而不加以控制,其残留物则会转移到食品中,造成人和动物的中毒,从而危害人类健康,造成环境污染[4-5]。因此,国家标准GB 2763-2019«食品安全国家标准 食品中农药最大残留限量»对不同蔬菜中抗蚜威农药的限量有相应的规定。
目前我国关于抗蚜威农药残留量的测定方法主要有气相色谱法[6]、气相色谱-质谱法[7]、高效液相色谱-质谱法[8-10]、柱后衍生-附荧光检测器的高效液相色谱法[11]、超临界流体色谱法[12]。其中,液相色谱-质谱仪和气相色谱-质谱仪价格昂贵,成本过高,不适合在各级地方实验室普及;气相色谱法需要在高温高压的条件下将目标物气化,再进入色谱柱进行分离,而抗蚜威农药有一定的热不稳定性,在高温碱性条件下会分解,因此气相色谱法也有一定的局限性。目前主流的检测方法和我国现行国家标准GB 23200.112-2018«食品安全国家标准 植物源性食品中9种氨基甲酸酯类农药及其代谢物残留量的测定 液相色谱-柱后衍生法»针对植物源性食品中9种氨基甲酸酯类农药及其代谢物残留的测定采用的都是液相色谱-柱后衍生化法,但是柱后衍生化法在从色谱柱到检测器的过程中还增加了衍生器,待测物需要通过漫长的通路才会进入荧光检测器进行检测,会导致仪器死体积过大、样品扩散,进而使色谱峰容易变宽、峰形变差,影响对目标物的定性和定量。并且,为保证衍生反应彻底以及定量结果的准确度,柱后衍生一般需要衍生试剂大量过载,因此还存在衍生试剂浪费的情况。目前常用的前处理方法有固相萃取法、液液萃取法、凝胶渗透色谱法和固相微萃取法等,这些前处理方法和QuEChERS 方法[13-18]相比,存在前处理效率低、耗时、成本过高的问题。
目前QuEChERS净化-柱前衍生结合高效液相色谱法测定烟叶中抗蚜威农药残留量的方法还未见报道,本工作着眼于此点进行研究。使用柱前衍生法替代以往普遍使用的柱后衍生法,待测物的极性降低,可以使用普通的非极性C18色谱柱进行分离,大大降低了成本,并且结合Qu ECh ERS前处理,降低了难度,缩短了时间,大大提高了工作效率。
1 试验部分
1.1 仪器与试剂
Agilent 1260II型高效液相色谱仪,配荧光检测器;Milli-Q academic型超纯水系统;FSH-II型高速电动匀浆器;TECHNE 型氮吹仪;HZQ/THZ型振荡器;AE100型电子分析天平。
0.05mol·L-1硼酸钠溶液:称取19.1 g十水合硼酸钠,完全溶解于1 L水中。
0.05mol·L-1氢氧化钠溶液:称取片状氢氧化钠1 g,用水溶解后转移至500 mL容量瓶中,定容,混合均匀,用0.45μm 滤膜过滤并脱气,滤液储存于棕色瓶中。
邻苯二甲醛(OPA)/2-巯基乙醇溶液:取50 mg OPA 于5 mL 甲醇中,缓慢旋转使其完全溶解后,转移至500 mL容量瓶中,用0.05 mol·L-1硼酸钠溶液稀释至刻度,混合均匀,用0.45μm 滤膜过滤并脱气。然后,加入0.5 mL 2-巯基乙醇,缓慢旋转直至混合均匀。一旦2-巯基乙醇加入后,切勿再将溶液脱气,将此溶液置于密闭的棕色玻璃瓶中,用铝箔包裹避光保存。
酸化水溶液(pH 3):用盐酸将500 mL 水酸化至pH 3,并用已校正的pH 计进行测定。
抗蚜威标准储备溶液:100 mg·L-1,准确移取1 000 mg·L-1抗蚜威标准溶液1 mL 置于10 mL容量瓶中,用乙腈定容,配制成质量浓度为100 mg·L-1的抗蚜威标准储备溶液。
抗蚜威标准溶液系列:分别移取100 mg·L-1的抗蚜威标准储备溶液10,100,200,500,1 000μL置于5支100 mL容量瓶中,用酸化水溶液(pH 3)稀释至刻度,混匀,配制成质量浓度为0.01,0.10,0.20,0.50,1.00 mg·L-1的抗蚜威标准溶液系列。储存于棕色玻璃瓶中,充氮气,除湿隔绝空气,于冷冻装置中密闭保存,此溶液至少稳定3个月。
抗蚜威标准溶液,质量浓度为1 000 mg·L-1;N-丙基乙二胺(PSA)、石墨化碳黑(GCB)、十八烷基键合硅胶(C18)固相吸附剂;OPA 的纯度不小于97%;2-巯基乙醇的纯度不小于98%;甲醇、乙腈均为色谱纯;片状氢氧化钠、氯化钠、无水硫酸镁(MgSO4)、十水合硼酸钠和盐酸均为分析纯;试验用水为超纯水(电阻率为18.2 MΩ·cm)。
样品来自市售烟叶。
1.2 仪器工作条件
EXTEND-C18色谱柱(250 mm×4.6 mm,5μm);柱温30 ℃;进样体积400 μL;流量1.0 mL·min-1;荧光检测器,激发波长339 nm,发射波长445 nm;增益10;流动相A 为水,B为甲醇,C 为乙腈;梯度洗 脱程序:0~5.3 min 时,A 为88%,B 为12%,C 为0;5.3~14.0 min 时,A 由88%降 至68%,B 由12%升 至16%,C 由0 升至16%;14.0~18.0 min时,A 由68%降至50%,B由16%升至25%,C 由16%升至25%,保持2.0 min;20.0~30.0 min时,A 由50%升至88%,B 由25%降至12%,C由25%降至0。
1.3 试验方法
1.3.1 样品提取
称取10.00 g均质完毕的烟叶样品置于50 mL离心管中,加入含1%(体积分数,下同)乙酸的乙腈溶液10 mL和2~3 g氯化钠后,振荡提取10 min,以转速9 500 r·min-1离心5 min。
1.3.2 Qu ECh ERS净 化
取上清液5 mL置于预先加有200 mg PSA 和900 mg MgSO4的离心管中,涡旋提取15 s,以转速9 500 r·min-1离心5 min,取上层提取液2.0 mL置于试管中,于35 ℃氮吹至近干。
1.3.3 柱前衍生
取1.3.2 节中试管,加入酸化水溶液(pH 3)500 μL、OPA/2-巯基乙 醇溶液 250 μL 和0.05 mol·L-1氢氧化钠溶液250μL,涡旋1 min溶解,经0.2μm 微孔膜过滤于进样小瓶中,用酸化水溶液(pH 3)定容至1 mL,于80 ℃水浴避光加热30 min,衍生完成,按照仪器工作条件测定。
2 结果与讨论
2.1 色谱柱的选择
氨基甲酸酯类农药没有特征光谱吸收基团,在碱性条件下可以与OPA/2-巯基乙醇溶液反应生成具有强荧光吸收的异氮(杂)茚基衍生物,进而降低待测物的极性,增大离子的电离效率,改善色谱分离度,提高检测器响应[19]。由于待测物的极性降低,可以使用普通的非极性C18色谱柱进行色谱分离,试验考察了BEH-C18色谱柱(250 mm×4.6 mm,5μm)、T3 色谱柱(250 mm×4.6 mm,5μm)和EXTEND-C18色谱柱(250 mm×4.6 mm,5μm)对目标物分离效果的影响。结果表明:使用T3色谱柱分离后,抗蚜威色谱峰存在拖尾,峰形较差;与BEH-C18色谱柱相比,使用EXTEND-C18色谱柱分离后,抗蚜威色谱峰尖锐、对称,分离效果更好。因此,试验选择EXTEND-C18色谱柱进行色谱分离。在所选定的仪器工作条件下,抗蚜威标准溶液和空白烟叶基质的色谱图见图2。
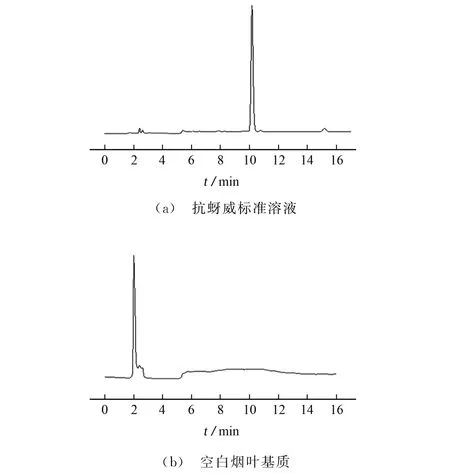
图2 典型色谱图Fig.2 Typical chromatograms
2.2 提取溶剂的选择
目前果蔬中农药的提取多是采用甲醇、乙腈或是酸性的甲醇溶液和乙腈溶液。试验以烟叶为空白基质,加标量为1 mg·kg-1,分别考察了提取溶剂乙腈、甲醇、含1%乙酸的甲醇溶液、含1%乙酸的乙腈溶液的提取效果。结果表明:甲醇和含1%乙酸的甲醇溶液的提取效率不如乙腈和含1%乙酸的乙腈溶液的效果好,根据试验经验分析,相较于乙腈,甲醇与水的互溶性更强,有相当大一部分水残留在有机相中,增大了共提取物的含量,即使在后续处理中加入盐(氯化钠),其分层效果也不如乙腈,甚至无法分层,而盐的加入能够使乙腈有机相和水相明显分离;含1%乙酸的乙腈溶液比乙腈的提取效果略好,是因为酸化的乙腈溶液形成了弱酸性的缓冲提取溶剂体系,能够一定程度地提高回收率,氨基甲酸酯类农药的平均回收率达到92.8%。因此,试验以含1%乙酸的乙腈溶液为提取溶剂。
2.3 QuECh ERS方法的优化
目前QuEChERS中用到的吸附剂填料主要有PSA、C18、GCB 和MgSO4。PSA 可通过弱阴离子交换、极性相互作用,吸附极性强的脂肪酸类和酚类干扰物;而C18则相反,通过非极性相互作用,吸附极性较弱的色素、维生素E、油脂、烯烃类、甾类干扰物;GCB主要用于吸附样品中的色素;MgSO4主要用于吸附样品中的水分。试验选取质量分数为1 mg·kg-1的加标烟叶样品为研究对象,按照1.3节中所述的前处理方法提取和衍生,考察了在4种吸附剂组合(分别为Ⅰ.PSA 200 mg+MgSO4900 mg;Ⅱ.GCB 15 mg+PSA 200 mg+MgSO4900 mg;Ⅲ.PSA 150 mg+C1850 mg+MgSO4900 mg;Ⅳ.GCB 15 mg+PSA 150 mg+C1850 mg+MgSO4900 mg)净化下加标烟叶样品中抗蚜威的回收率,结果见图3。
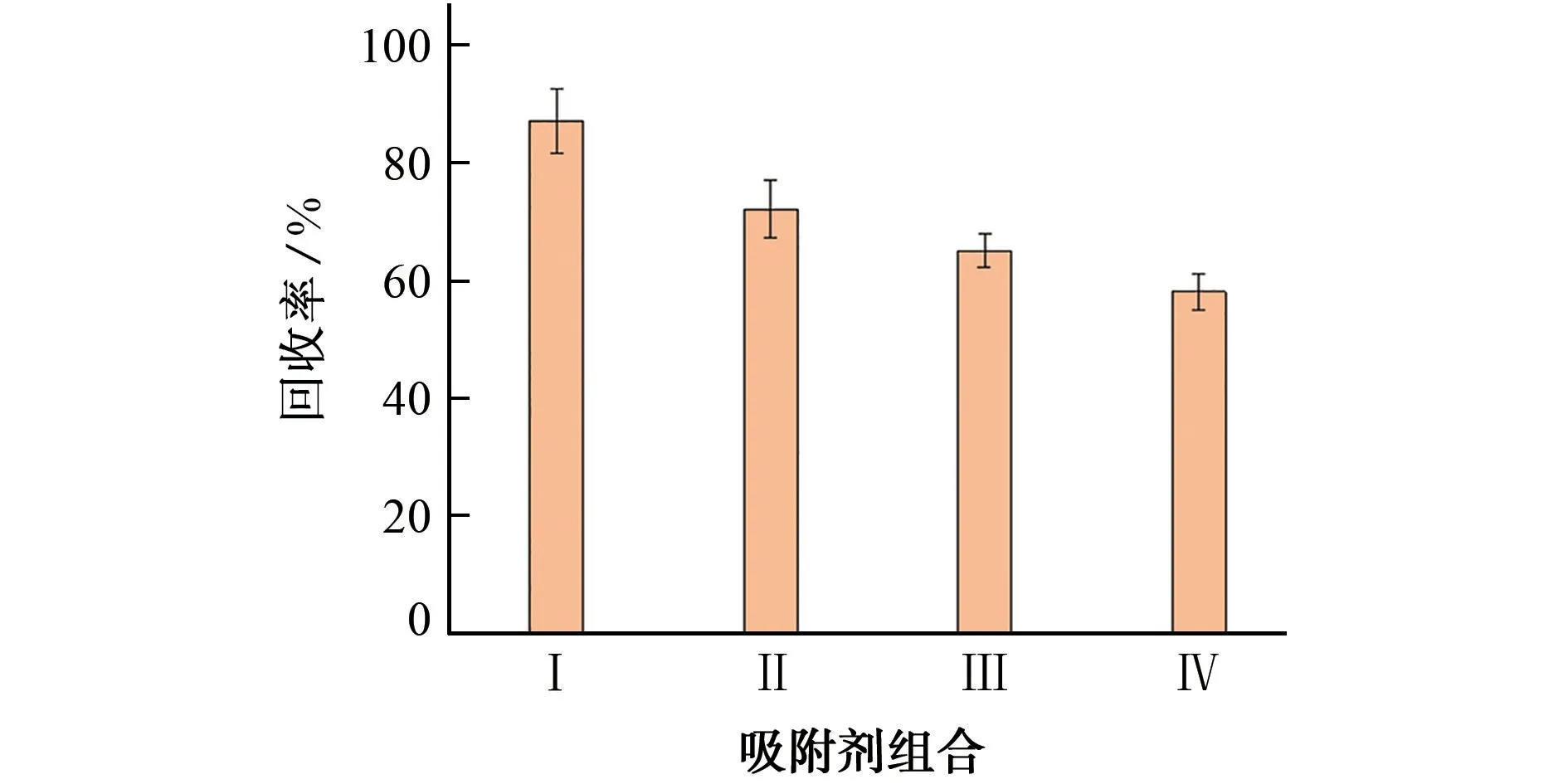
图3 在4种吸附剂组合净化下加标烟叶样品中抗蚜威的回收率Fig.3 Recovery of pirimicarb in the spiked sample of tobacco leaf purified with 4 adsorbent combinations
结果表明:在吸附剂组合I净化下,加标烟叶样品中抗蚜威的回收率最高,这是因为GCB对抗蚜威农药有一定的吸附作用;虽然C18在日常检测中主要用于吸附脂类物质,但烟叶中脂类物质较少,导致C18对抗蚜威农药产生了吸附,回收率降低。在吸附剂组合Ⅳ净化下,加标烟叶样品中抗蚜威的回收率最低,是GCB和C18共同对氨基甲酸酯类农药的吸附而导致的结果,与文献[20]研究结果一致。因此,试验优选组合Ⅰ作为净化吸附剂。
2.4 衍生条件的选择
2.4.1 0.05 mol·L-1氢氧化钠溶液的添加量
在不加氢氧化钠溶液时,衍生反应无法发生。固定衍生反应时间、温度和衍生试剂添加量,试验采用0.05 mol·L-1氢氧化钠溶液作为水解试剂,考察了其添加量(50,100,150,200,250,300μL)对抗蚜威回收率的影响,结果见图4。
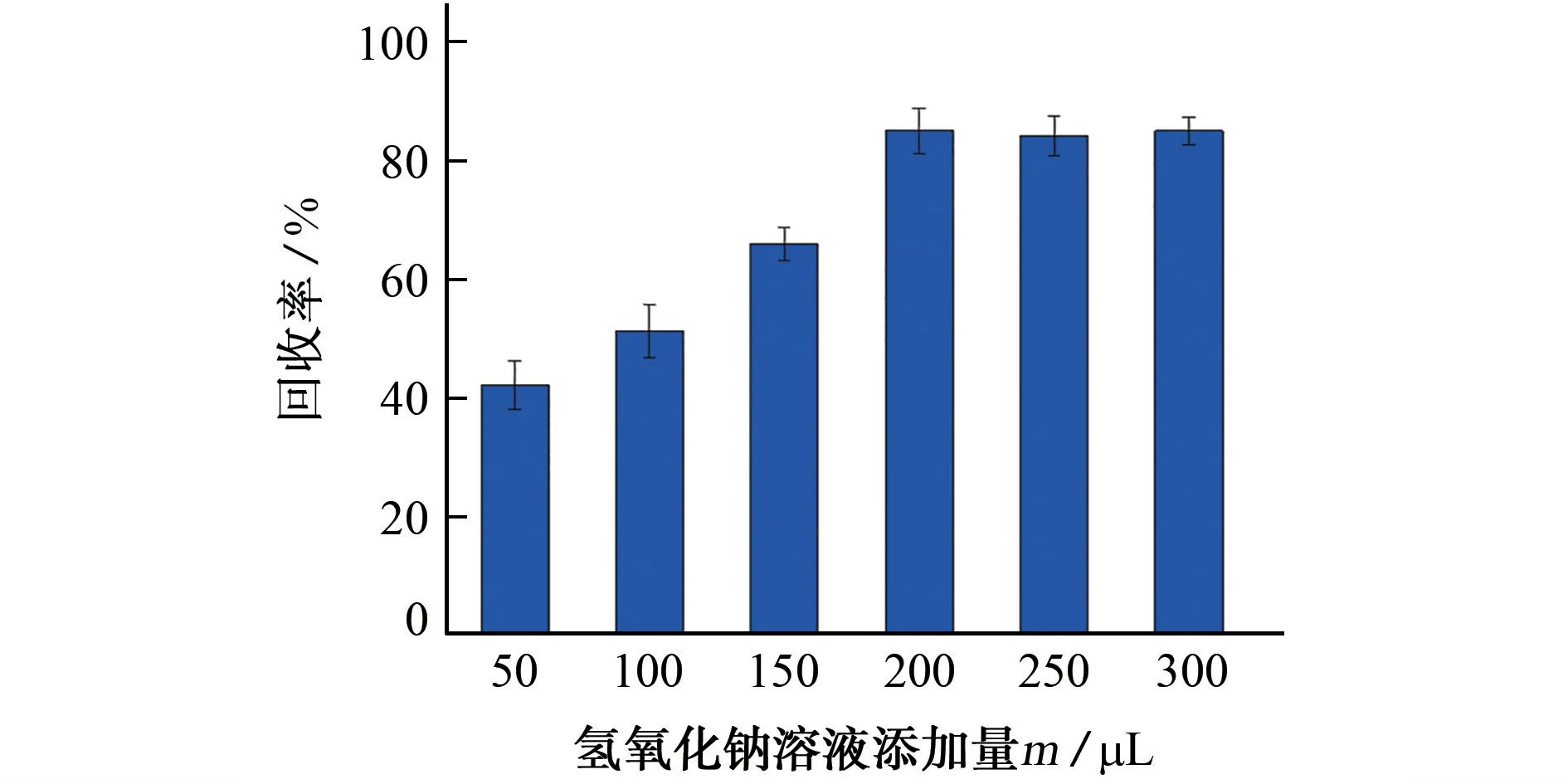
图4 氢氧化钠溶液添加量对抗蚜威回收率的影响Fig.4 Effect of addition amount of sodium hydroxide solution on recovery of pirimicarb
由图4可知:当0.05 mol·L-1氢氧化钠溶液添加量达到200μL时,抗蚜威回收率接近最大,衍生效果接近最大;当0.05 mol·L-1氢氧化钠溶液添加量达到250μL时,抗蚜威回收率稳定;继续增大0.05 mol·L-1氢氧化钠溶液添加量,抗蚜威回收率不再改变。综合考虑,试验优 选250 μL 为0.05 mol·L-1氢氧化钠溶液的添加量。
2.4.2 OPA/2-巯基乙醇溶液的添加量
由于抗蚜威不具备典型的荧光和紫外吸收基团,因此需要进行衍生化反应使之具有荧光吸收的特征检验基团,方能在荧光检测器上有特征响应。固定衍生反应时间、温度和0.05 mol·L-1氢氧化钠溶液添加量,选用OPA/2-巯基乙醇溶液为衍生试剂,按照1.1节中所述方法配制,试验考察了不同衍生试剂添加量(50,100,150,200,250,300μL)的衍生效果,对比衍生后待测物的峰面积,计算抗蚜威的回收率,结果见图5。
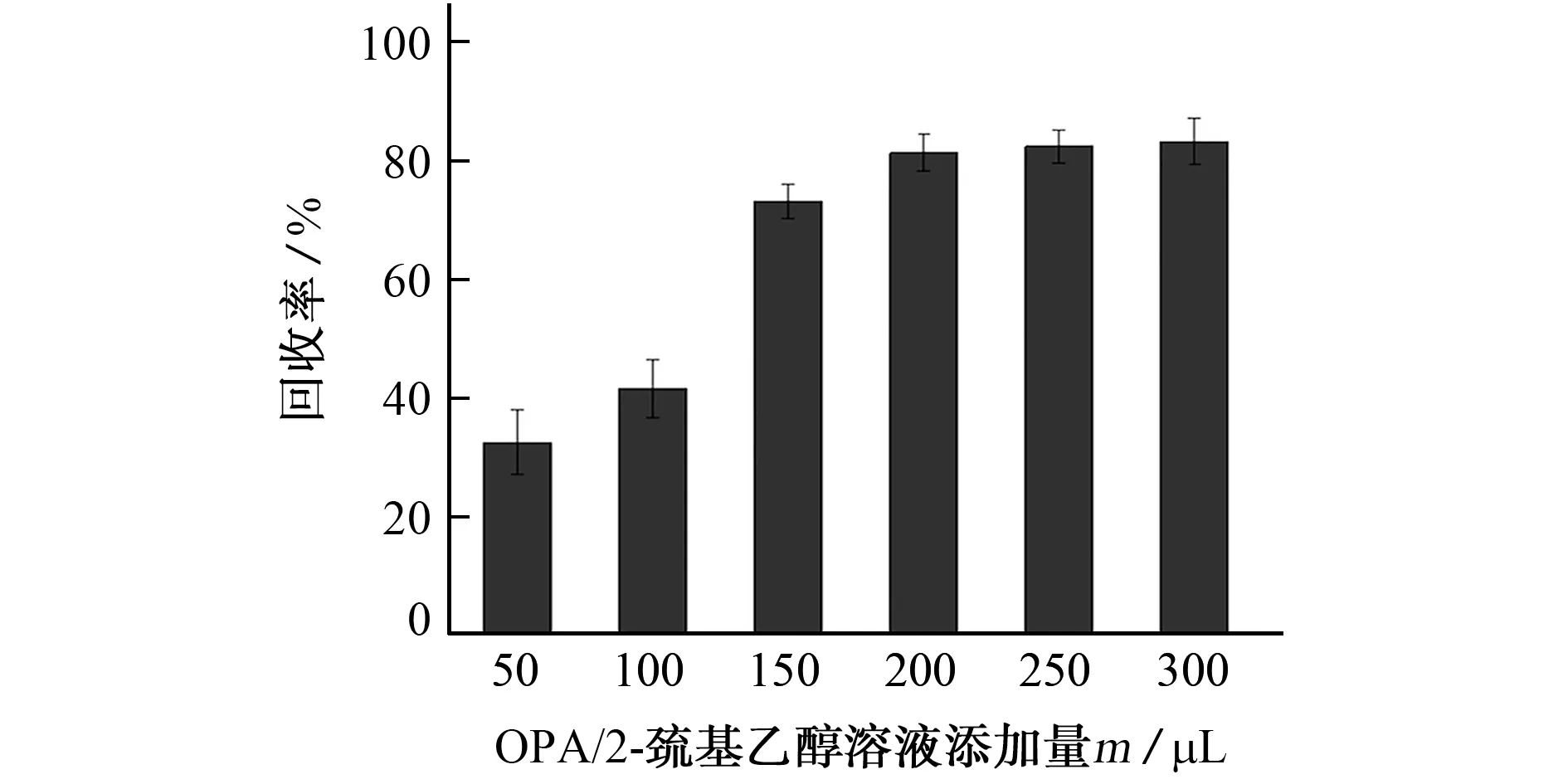
图5 OPA/2-巯基乙醇溶液添加量对抗蚜威回收率的影响Fig.5 Effect of addition amount of OPA/2-mercaptoethanol solution on recovery of pirimicarb
结果表明,OPA/2-巯基乙醇溶液添加量达到200μL 时,目标物衍生效果较好,抗蚜威回收率达到稳定。衍生化反应一般需要衍生试剂过量才能够保证衍生反应完全,试验为保证衍生化反应充分,遵循定量准确和节约试剂用量的原则,最终选择250μL为OPA/2-巯基乙醇溶液的添加量。
2.5 标准曲线、检出限和测定下限
按照仪器工作条件测定抗蚜威标准溶液系列,以抗蚜威的质量浓度为横坐标,对应的衍生物峰面积为纵坐标绘制标准曲线。结果表明,抗蚜威标准曲线的线性范围为0.01~1.00 mg·L-1,线性回归方程为y=1.174×102x-2.430×10-1,相关系数为0.999 8。
以3倍信噪比(S/N)确定检出限(3S/N),以10倍信噪比确定测定下限(10S/N),结果得出检出限为0.01 mg·kg-1,测定下限为0.05 mg·kg-1。
2.6 精密度和回收试验
按照试验方法对阴性烟叶样品进行0.05,0.25,0.50 mg·kg-1等3个浓度水平(按照试验方法处理后,加标量相当于0.01,0.50,1.00 mg·L-1)的加标回收试验,每个加标样品平行测定6次,并计算回收率和测定值的相对标准偏差(RSD),结果见表1。
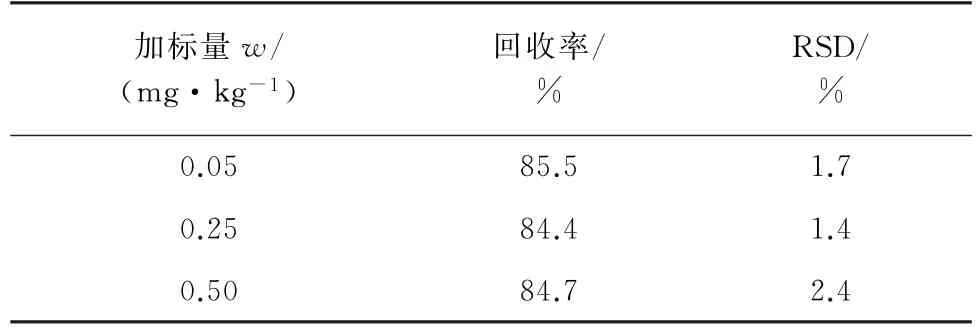
表1 精密度和回收试验结果(n=6)Tab.1 Results of tests for precision and recovery(n=6)
由表1可知,回收率为84.4%~85.5%,测定值的RSD 为1.4%~2.4%,表明该方法的精密度和重现性良好,可以满足GB/T 27404-2008«实验室质量控制规范 食品理化检测»中有关多残留测定的要求。
2.7 样品分析
从贵阳市农贸市场随机购买30份烟叶样品,按照1.3 节试验方法前处理,1.2 节仪器工作条件测定,结果显示,有6 份烟叶样品检出抗蚜威,其余24份烟叶样品均未检出,检出率为20%,其中最高检出量为0.06 mg·kg-1。
本工作提出了QuEChERS净化-柱前衍生法结合高效液相色谱法测定烟叶中抗蚜威残留量的方法,相较于传统的检测方法,能够避免柱后衍生仪的使用,大大降低试验成本,简化了前处理步骤。该方法重现性好、准确度高、线性关系良好,可满足烟叶中抗蚜威的快速测定。
猜你喜欢
煤化工(2022年3期)2022-07-08
初中生学习指导·中考版(2021年2期)2021-09-10
色谱(2021年7期)2021-06-07
西南农业学报(2020年9期)2020-12-10
首都食品与医药(2020年1期)2020-10-21
活力(2019年15期)2019-09-25
山东工业技术(2016年10期)2016-09-06
中学生数理化·中考版(2015年12期)2015-09-10
作物研究(2014年6期)2014-03-01
中国烟草学报(2012年1期)2012-04-09
