
一种氨基哌嗪功能化的丹磺酰氯荧光衍生试剂的合成及其在高效液相色谱法定量分析微藻油脂中的应用
2022-06-23 14:42:10郭子娴全红花
理化检验-化学分册 2022年5期
郭子娴,蒋 妍,全红花,李 明
(扬州大学 环境科学与工程学院,扬州 225127)
微藻是一种广泛分布于海洋、淡水中的单细胞生物[1],其中富含的油脂是重要的生物质燃料[2],不同种类微藻中油脂的组成及含量相差较大[3-6]。目前,相关测定方法有衍生化-气相色谱法、近红外光谱法、毛细管电泳法、高效液相色谱法。衍生化-气相色谱法对热敏感物质影响较大[7],近红外光谱法操作较难以及所需样品量较大[8],毛细管电泳法的表面电渗变化对分离重现性影响较大[9],而附荧光检测器的高效液相色谱法(FLD-HPLC)的分离度及灵敏度较高,适用于不同脂肪酸衍生物的分析[10-11]。脂肪酸由于不具有荧光特性,需要通过荧光衍生试剂与脂肪酸反应生成荧光衍生物来实现检测[12]。常见的荧光衍生试剂有溴甲基类、哌嗪类、重氮甲烷类、氨基类化合物等[13-14]。丹磺酰氯(DNS-CL)是常用的氨基化合物荧光衍生试剂的原料,具有灵敏度高的优点[15-17]。但DNS-CL 不能直接用于脂肪酸的衍生化。
鉴于此,本工作制备了二甲基氨基哌嗪功能化的DNS-CL 荧光衍生试剂(DNS-Pi-NH2),并用其衍生化小球藻中碳原子为10~20 的脂肪酸,用FLD-HPLC测定脂肪酸衍生物的含量,以期为微藻中碳原子数为10~20的脂肪酸含量的准确测定提供技术参考。
1 试验部分
1.1 仪器与试剂
L-2000型高效液相色谱仪;Bojin C18固相萃取柱(2 000 mg/12 mL);RE52CS型旋转蒸发仪;Cary 610/670型显微红外光谱仪;AVANCE 600型核磁共振波谱仪;ma Xis型超高分辨飞行时间质谱仪,配电喷雾离子(ESI)源。
单标准储备溶液:2.5×10-4mol·L-1,取适量各脂肪酸标准品,用N,N-二甲基甲酰胺(DMF)溶解并定容至10.0 mL容量瓶中,摇匀备用。
混合标准溶液系列:取适量脂肪酸单标准储备溶液,用DMF 逐级稀释,配制成2.0×10-10,2.0×10-9,2.0×10-8,2.0×10-7,2.0×10-6,2.0×10-5,2.0×10-4mol·L-1混合标准溶液系列。
十二烷酸、十四烷酸、十六烷酸、十八烷酸和二十烷酸标准品的纯度均大于98%;顺-9-十八(碳)烯酸、顺-9,12-十八(碳)烯酸、顺-7,10,13-十六(碳)烯酸标准品的纯度均大于85%;硬脂酸甲酯标准品的纯度大于97%;DMF的纯度大于99.5%;DNS-CL、N-(2-氨乙基)哌嗪的纯度均大于99%;三苯基膦(TPP)的纯度大于95%;二丙基二硫醚(DPDS)的纯度大于98.0%;试验用水为去离子水。
小球藻(微藻的一种)脂肪酸甲酯样品由韩国浦项科技大学微流控应用化学中心提供。
1.2 仪器工作条件
Eclipse XDB C8色谱柱(250 mm×4.6 mm,5μm);流动相为90%(体积分数,下同)乙腈溶液;等度洗脱,时间30 min;流量0.8 mL·min-1;进样量10μL;荧光激发(λex)和发射波长(λem)分别为360,520 nm。
1.3 试验方法
1.3.1 DNS-Pi-NH2的合成
取DNS-CL 100 mg,在避光条件下溶于5 mL丙酮中,得到A 液。取碳酸钠30 mg和N-(2-氨乙基)哌嗪300 mg,溶于3 mL水中,然后与10 mL乙腈混合,得到B液。将A 液缓缓滴入B 液中,避光搅拌30 min,得到C液。加入和C液等体积的乙酸乙酯进行萃取,摇匀后静置分层。收集上层(乙酸乙酯层)液体,于78 ℃旋转蒸发至溶液体积不大于1 mL。将得到的液体过C18固相萃取柱。洗脱液用红外光谱、核磁共振碳谱(13C NMR)以及ESI-超高分辨飞行时间质谱(ESI-MS,质量数362.286 6)表征,所得物质即为DNS-Pi-NH2。
1.3.2 脂肪酸标准溶液的衍生化
取上述DNS-Pi-NH2和0.039 66 g TPP,分别用1.0 mL乙腈溶解制得DNS-Pi-NH2、TPP 溶液。取脂肪酸混合标准溶液、DNS-Pi-NH2溶液、DPDS和TPP溶液各175,10,25,25μL于1.5 mL塑料离心管中,室温振荡10 min,使衍生反应充分进行。待反应完成后,将产物转移至10 mL 容量瓶中,用90%乙腈溶液稀释至刻度,供FLD-HPLC分析。
1.3.3 样品的测定
将含0.5 mol·L-1氢氧化钾的乙醇溶液5 mL置于水浴烧杯中,加入2.0 mg小球藻脂肪酸甲酯样品。连接回流冷凝管,于79 ℃加热60 min,直至烧杯内溶液澄清透明,无明显油珠,此时小球藻脂肪酸甲酯已被完全皂化。取下水浴烧杯,加入2~3滴酚酞指示剂,滴加0.5 mol·L-1盐酸溶液至红色消失。继续于79 ℃水浴加热30 min,使乙醇完全蒸发。所得固体用10 mL 水清洗,以转速8 000 r·min-1离心5 min。再加入适量水于离心管中,振荡5 min,产物用0.45μm 尼龙注射器过滤,以去除氯化钾等杂质,烘干后,即得小球藻脂肪酸样品。取1 mg小球藻脂肪酸样品,按照1.3.2节试验方法衍生和测定。
2 结果与讨论
2.1 DNS-Pi-NH2 表征结果的分析
2.1.1 红外光谱
所得红外光谱图见图1。
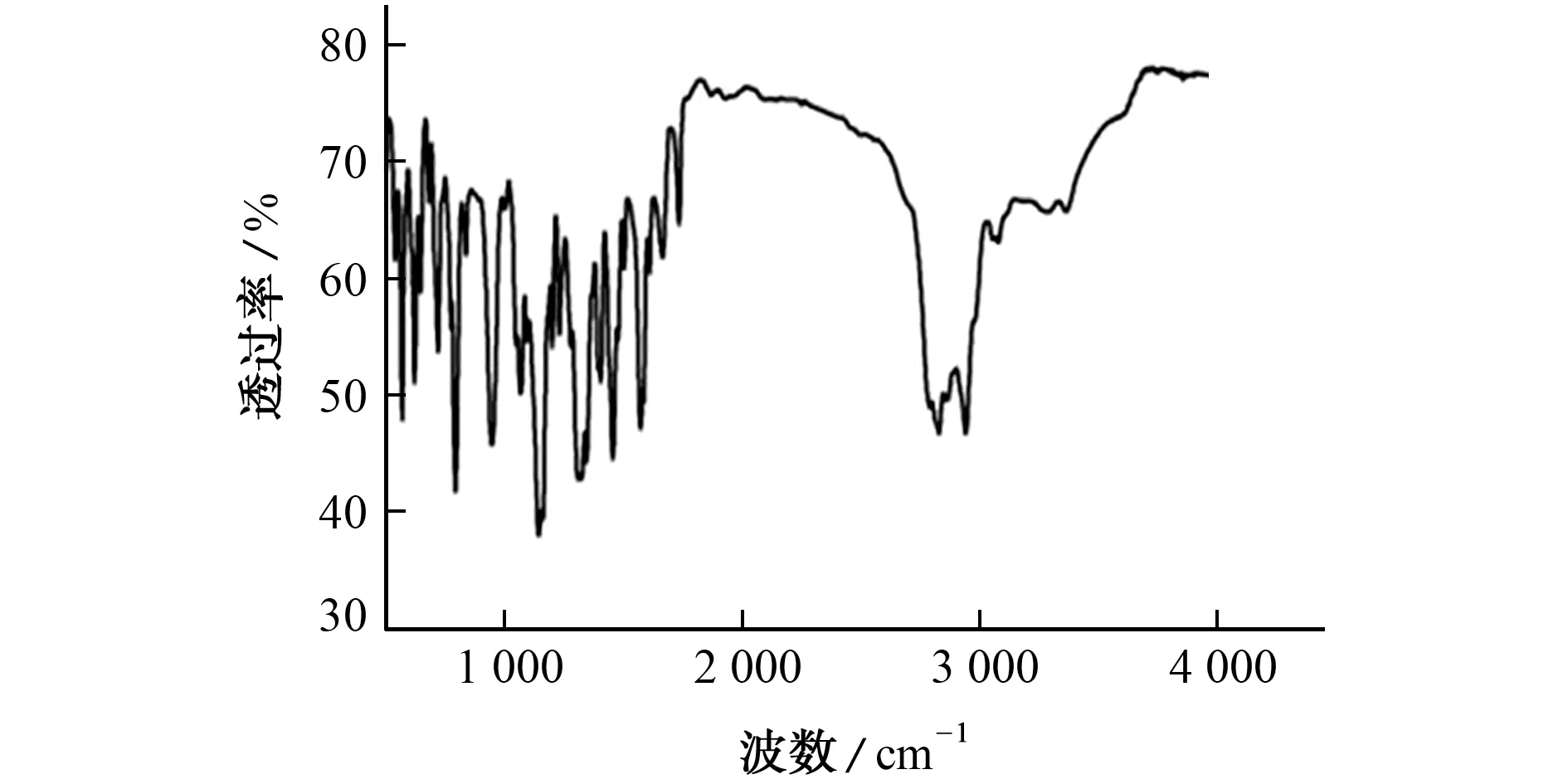
图1 红外光谱图Fig.1 Infrared spectrum
参考文献[18],分析图1中官能团的红外特征峰:3 363.4 cm-1处为N-H 的伸缩振动吸收峰,2 914.1,2 828.0 cm-1处为C-H 的伸缩振动吸收峰,1 455.3 cm-1处为CH2的变形振动吸收峰;1 665.3 cm-1处为芳环中C=C 骨架伸缩振动吸收峰,说明合成的物质为芳香族化合物;1 572.8 cm-1处为N-H 的变形振动吸收峰,1 455.3 cm-1处为芳环C=C 伸缩振动和CH3的C-H 不对称变形振动的重合吸收峰,1 323.2 cm-1处为C-N(芳基碳)的伸缩振动吸收峰,1 143.5,1 067.5 cm-1处为C-N(烷基碳)的伸缩振动吸收峰,945.8 cm-1处为N(CH3)2的弯曲振动吸收峰。根据以上推断,得出合成物质中含有苯环、N-H、C-N(芳基碳)、C-N(烷基碳)、CH2、CH3、N(CH3)2等官能团。
2.1.213C NMR
合成目标物的13C NMR 图见图2。
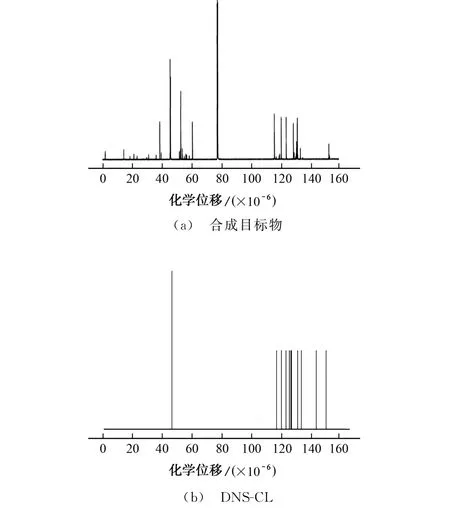
图2 合成目标物和DNS-CL的13 C NMR 图Fig.2 13 C NMR images of synthetic target and DNS-CL
结果表明:大于110×10-6的峰归属于DNSCL芳环中的碳;30×10-6~70×10-6的4个主峰归属于2-乙基氨基哌嗪中的碳;77×10-6处峰归属于溶剂氘代三氯甲烷中的碳。综上可说明DNS-Pi-NH2已成功制备,推测其结构见图3。
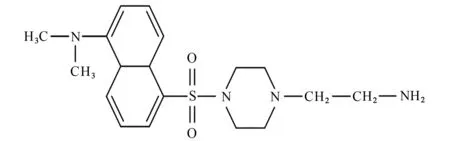
图3 DNS-Pi-NH2 的结构Fig.3 Structure of DNS-Pi-NH2
2.1.3 ESI-MS
为了明确合成目标物(化学式M)的结构,对该物质进行ESI-MS分析,所得准分子离子[M+H]+的质量数为363.185 4,和DNS-Pi-NH2的理论值一致,说明DNS-Pi-NH2已正确合成。
2.2 色谱条件的选择
2.2.1 色谱柱
以1.0×10-6mol·L-1的十二烷酸、十六烷酸、十八烷酸、二十烷酸混合标准溶液为待测对象,考察了Eclipse XDB C8色谱柱和Hypersil ODS C18色谱柱对4种典型脂肪酸衍生物的分离效果的影响。结果显示:以Hypersil ODS C18色谱柱分离时,十八烷酸和二十烷酸不能实现基线分离;Eclipse XDB C8色谱柱可实现4种目标物基线分离,且峰形较好。因此,试验选择以Eclipse XDB C8色谱柱分离各脂肪酸衍生物。
2.2.2 流动相乙腈溶液体积分数
试验进一步考察了流动相乙腈溶液的体积分数分别为50%,90%时对以上4种脂肪酸衍生物分离效果的影响,结果见图4。
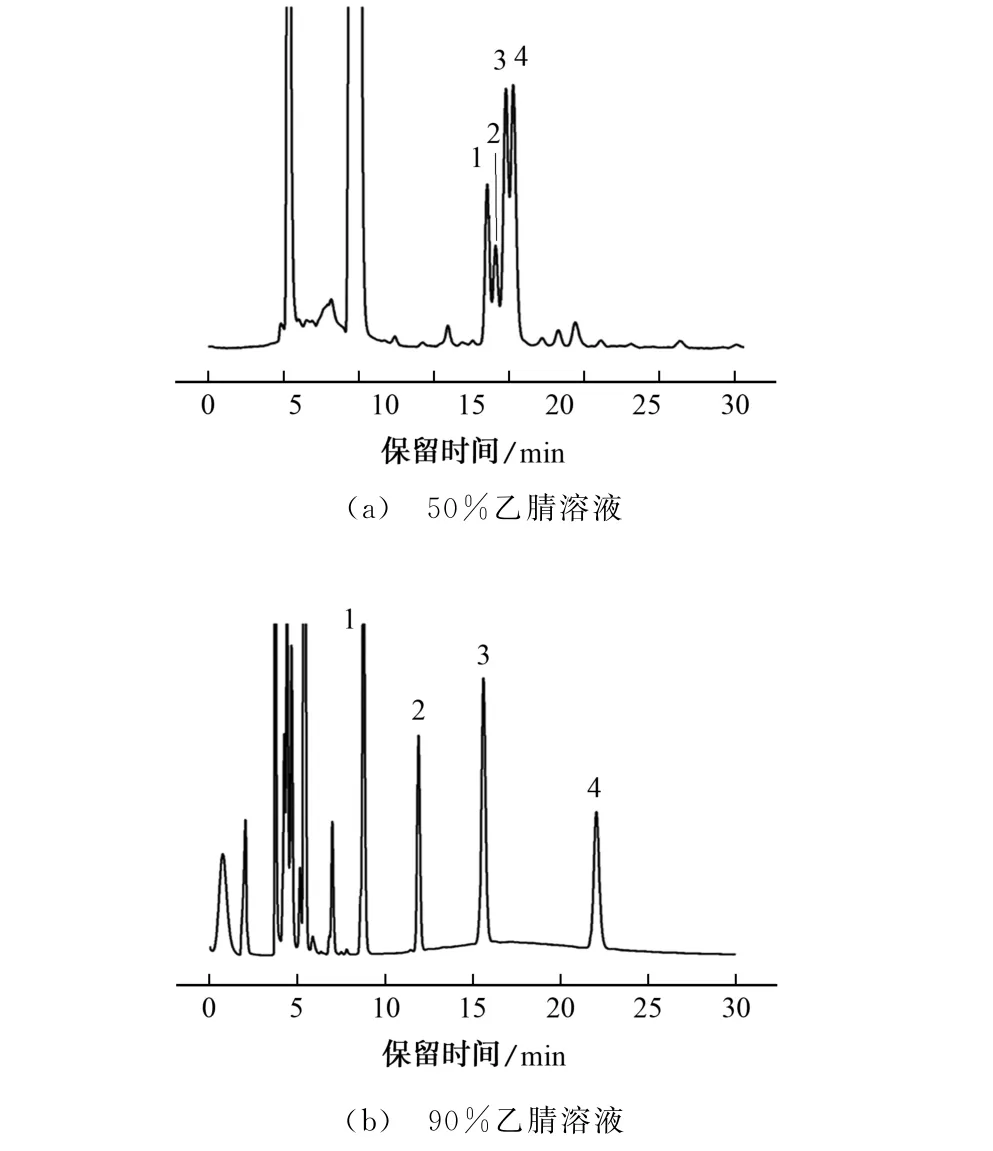
图4 不同体积分数乙腈溶液下4种脂肪酸衍生物的色谱图Fig.4 Chromatograms of four fatty acid derivatives under different volume fractions of acetonitrile solution
由图4可知:当乙腈溶液体积分数为50%时,可在色谱图中明显观察到4 种脂肪酸衍生物,但4种脂肪酸衍生物色谱峰重叠严重,分离度差,未能得到基线分离;当乙腈溶液体积分数为90%时,30 min内可实现4 种脂肪酸衍生物的基线分离。因此,试验选择的流动相为90%乙腈溶液。
在上述优化试验条件下,1.0×10-6mol·L-1混合标准溶液的色谱图见图5。
由图5可知,8种脂肪酸衍生物可在30 min内完成分离。
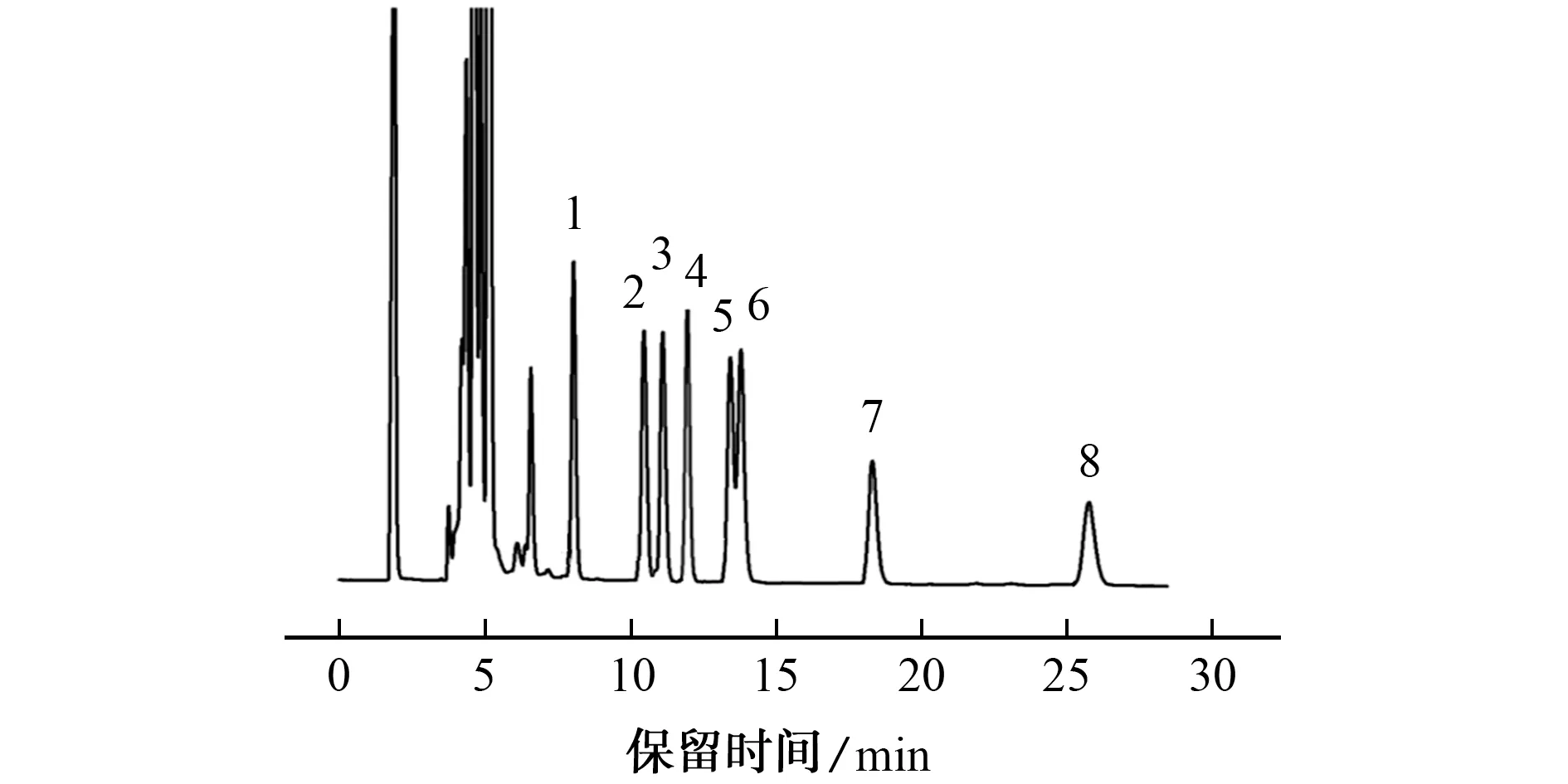
图5 混合标准溶液的色谱图Fig.5 Chromatogram of the mixed standard solution
2.3 方法学验证
2.3.1 标准曲线和检出限
按照1.3.2 节试验方法分析混合标准溶液系列,以各脂肪酸的浓度为横坐标,其对应的峰面积为纵坐标绘制标准曲线。结果显示,各脂肪酸标准曲线的线性范围均为2.0×10-10~2.0×10-4mol·L-1,线性回归方程、相关系数见表1。
按照1.3.2节试验方法对1.0×10-10mol·L-1脂肪酸混合标准溶液重复分析11次,计算峰面积的标准偏差(s),以3s与标准曲线斜率(k)的比值计算检出限(3s/k),所得结果见表1。
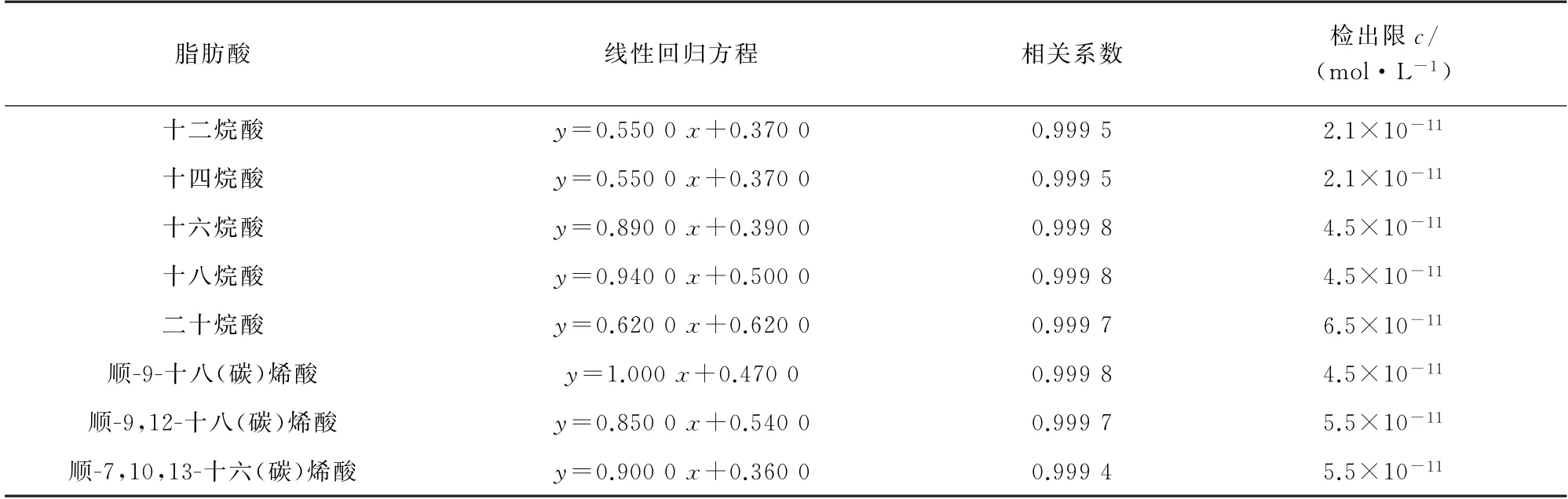
表1 线性参数和检出限Tab.1 Linearity parameters and detection limits
2.3.2 精密度和回收试验
用微藻培养液进行精密度和准确度试验。向该微藻培养液中添加适量混合标准溶液,使加标量达到2.0×10-6mol·L-1,再重复测定5次,计算回收率和测定值的相对标准偏差(RSD)。结果显示,各脂肪酸的回收率为95.3%~102%,测定值的RSD为1.7%~2.6%。
2.4 样品分析
按照试验方法分析小球藻脂肪酸甲酯样品,色谱图见图6。
由图6可知,小球藻样品中含有的脂肪酸依次为十六烷酸、顺-9,12-十八(碳)烯酸、顺-7,10,13-十六(碳)烯酸、顺-9-十八(碳)烯酸,与文献结果一致[19],4种脂肪酸检出量分别为3.08,0.64,0.83,2.57 mg·g-1。
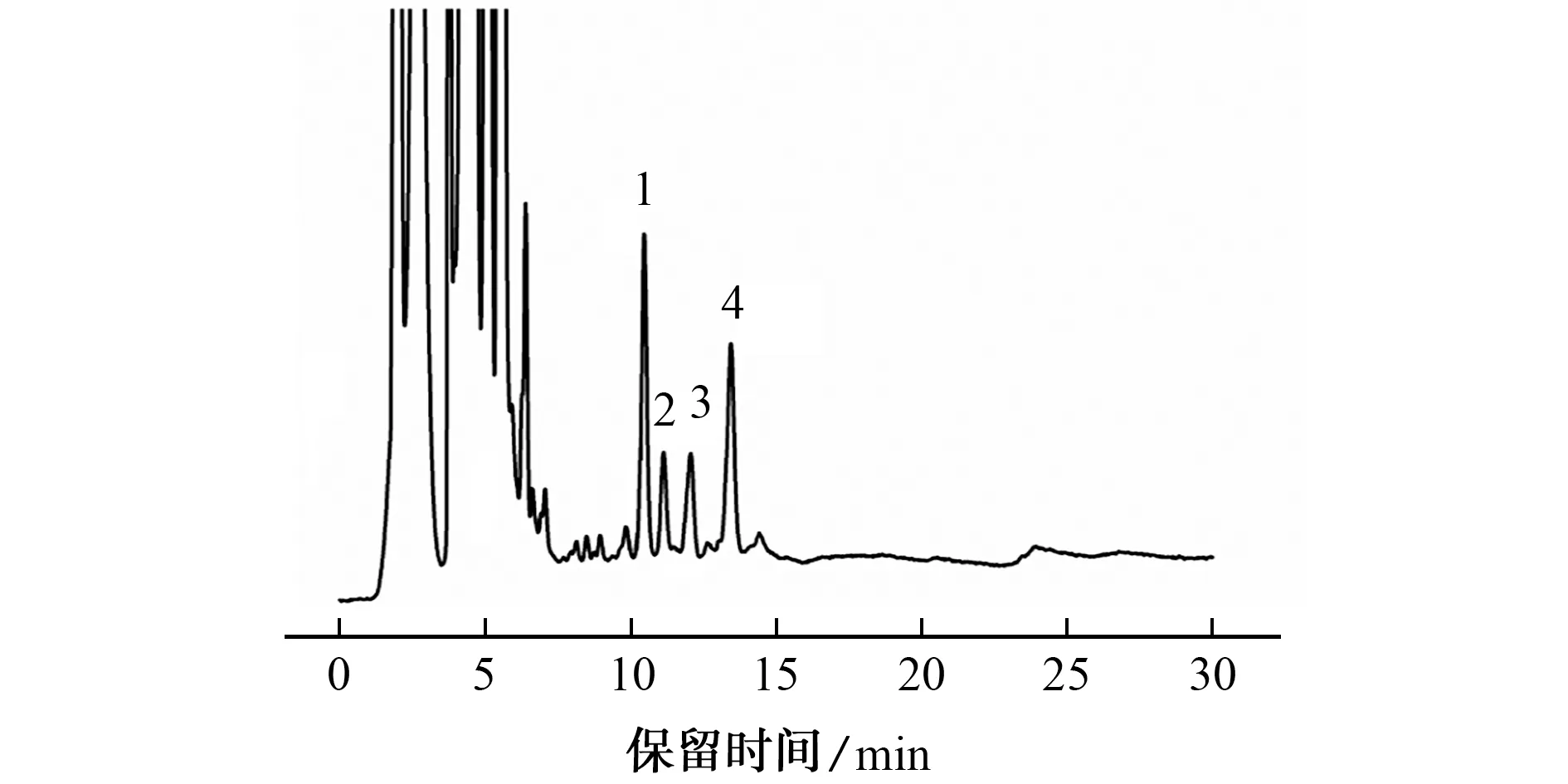
图6 实际样品的色谱图Fig.6 Chromatogram of the actual sample
本工作以自制DNS-Pi-NH2衍生化碳原子为10~20的脂肪酸,用FLD-HPLC 检测。本方法衍生温度低、反应时间短、样品用量少、精密度和准确度较好,可为微藻中脂肪酸含量的测定提供一种简便、快速、有效的分析方法。
猜你喜欢
古今农业(2022年1期)2022-05-05 06:58:42
陶瓷学报(2019年5期)2019-01-12 09:17:44
环境保护与循环经济(2017年3期)2017-09-26 11:42:30
电源技术(2016年2期)2016-02-27 09:04:55
中国塑料(2015年6期)2015-11-13 03:03:11
中国医药科学(2015年5期)2015-08-01 14:14:51
食品工业科技(2014年23期)2014-03-11 18:19:31
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:31
色谱(2013年6期)2013-07-13 05:24:12
中国工程咨询(2013年3期)2013-02-13 02:50:20
