
ABCC8基因复合杂合突变致儿童KATP-HI 9例临床分析
2022-06-17 07:48曾俏徐子迪张琳吴玉筠刘敏闫洁桑艳梅朱逞倪桂臣
疑难病杂志 2022年6期
曾俏,徐子迪,张琳,吴玉筠,刘敏,闫洁,桑艳梅,朱逞,倪桂臣
先天性高胰岛素血症(congenital hyperinsulinism,CHI)是新生儿和儿童持续复发性低血糖常见的原因之一,是一种因胰岛素分泌调节异常引起的遗传异质性疾病。随着研究的不断进展,迄今已发现了15种CHI相关的致病基因,主要包括ABCC8、KCNJ11、GLUD1等,相应地构成14种遗传学类型。其中,ATP敏感钾通道型先天性高胰岛素血症(adenosine triphosphate-sensitive potassium channel hyperinsulinism,KATP-HI)是CHI最常见的类型,是因ABCC8和KCNJ11基因失活突变导致[1],这2个基因分别编码磺脲类受体1蛋白(sulfonylurea receptor1,SUR1)和内向整流钾通道蛋白(potassium inward rectifying channel,Kir6.2)。迄今为止,ABCC8基因已发现了400多种突变,KCNJ11基因已发现了60多种突变[2]。其中,ABCC8基因复合杂合突变临床较为少见,迄今国内外报道的只有几十例。导致KATP-HI的KCNJ11复合杂合突变临床报道目前仅3例[3-5]。本研究搜集携带ABCC8复合杂合突变的9例KATP-HI 患儿为研究对象,对其临床特征及遗传学特征进行分析,以期提高临床医师对该病的认识。
1 临床资料
1.1 一般资料 选取2012年5月—2017年8月北京儿童医院内分泌遗传代谢科收治的携带ABCC8基因复合杂合突变KATP-HI患儿9例,均符合CHI的诊断标准。具体诊断标准如下[6]:(1)高胰岛素血症(血浆胰岛素>2 mIU/L);(2)低脂肪酸血症(血浆游离脂肪酸 <1.5 mmol/L);(3)低酮血症(血浆β-羟丁酸 <2.0 mmol/L);(4)1 mg静脉胰高血糖素试验反应:血糖变化>1.7 mmol/L。其中男6例,女3例。起病年龄生后1 d~2岁,中位起病年龄为生后1 d。出生体质量1.8~4.9 kg,正常出生体质量儿2例,巨大儿6例,低出生体质量儿1例,见表1。
1.2 临床特征 首发症状为无明显诱因抽搐3例,有低血糖症状3例(如反应弱、头面部青紫和四肢肌张力减低等),无症状性低血糖3例。1例患儿(例9)的姐姐患有CHI,于5岁时起病,与患儿携带相同突变。其余8例患儿无低血糖家族史。9例均无糖尿病家族史及母孕期糖耐量受损史。2例(例1和例4)曾分别于香港养和医院和复旦大学附属华山医院进行18-氟左旋多巴正电子发射断层(18F-L-DOPA PET/CT)胰腺扫描,胰腺组织学类型均提示弥漫型。
1.3 基因突变携带情况 9例患儿均携带ABCC8基因(NM_000352)复合杂合突变,见表1。例2的p.R1245G、p.N72K,例3的p.A471V、p.L1211P,例4的p.T1042QfsTer75、p.E945RfsTer25,例7的p.A640V、p.Q1196*及例9的p.P1161R突变为中国人首次报道。
1.4 治疗与转归 二氮嗪治疗8例,有效3例,无效5例。2例在应用二氮嗪治疗期间出现血小板减少、中性粒细胞绝对值减少、低血钾、纳差等不良反应。奥曲肽治疗4例,均有效,治疗过程中1例患儿出现肝功能损害。行胰腺次全切除术治疗者1例(例4)。经长期随访,3例通过少量多餐饮食维持血糖,空腹时间延长时,偶有低血糖发作;2例长期奥曲肽治疗,3例自行缓解,1例失访,见表1。
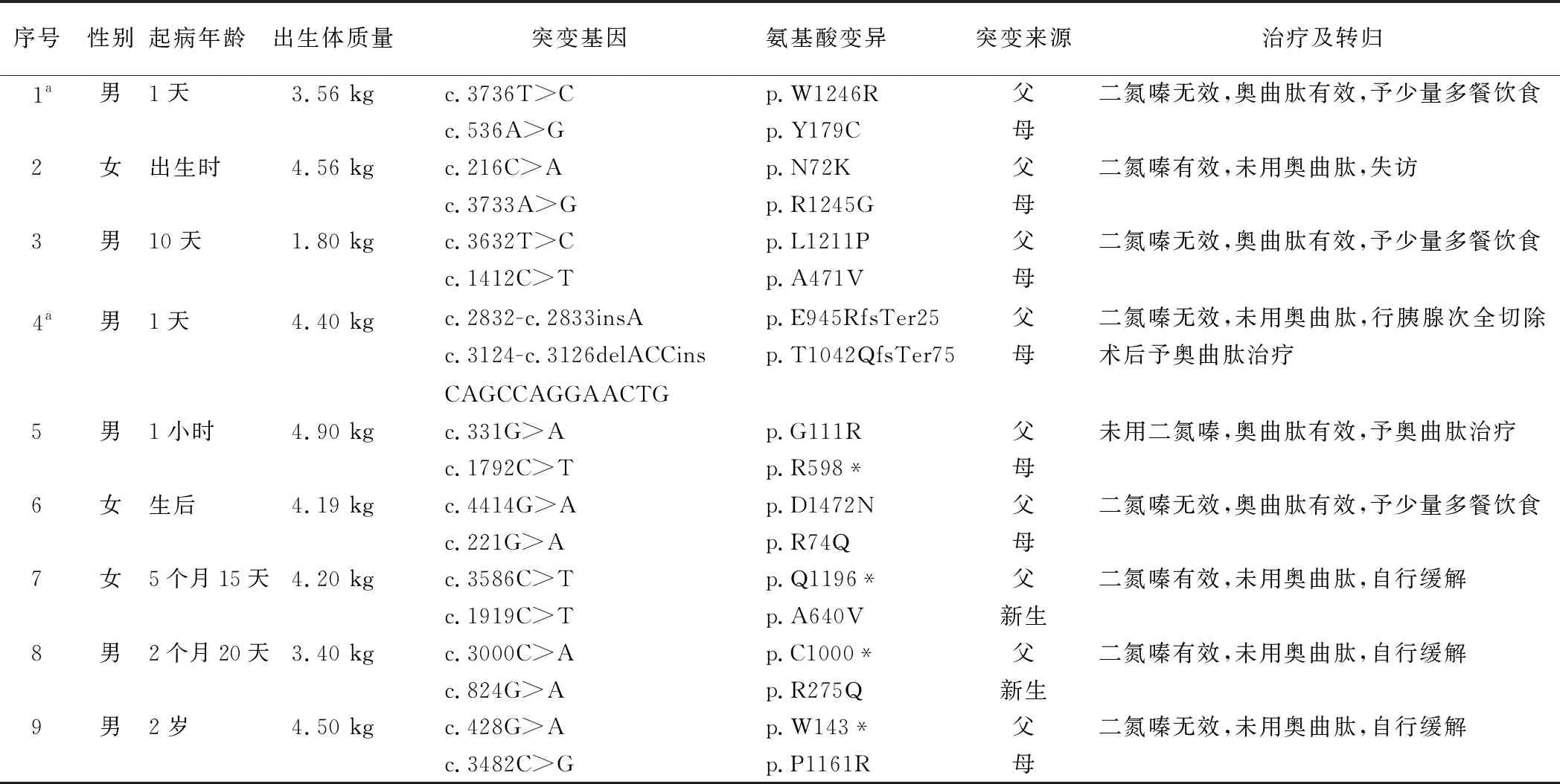
表1 9例KATP-HI患儿携带ABCC8基因复合杂合突变情况及主要临床特征
例4于1岁11个月时行胰腺次全切除术(90%),术后仍有低血糖发作,故予奥曲肽长期治疗维持正常血糖水平,目前4岁,奥曲肽剂量5 μg·kg-1·d-1。例9于5岁时自行缓解,随访过程中无低血糖发作,自2岁起诊断癫痫,服用抗癫痫药物至今,仍有间断抽搐发作,频率4~5次/年,每次持续约 2 min缓解。例3于3岁时自行停用奥曲肽,空腹时间延长时偶有低血糖及抽搐发作,发作频率约每2个月1次,口服葡萄糖水后1~2 min缓解,现4岁,语言发育迟缓。例5出院后奥曲肽治疗至今,纳差时出现低血糖发作(1.9~2.4 mmol/L),现3岁,语言发育迟缓。
2 讨 论
KATP-HI是CHI最常见和最严重的类型,其遗传方式多为常染色体隐性遗传,少数为常染色体显性遗传,还有一部分为新生突变。
ABCC8和/或KCNJ11基因复合杂合突变导致的KATP-HI临床较罕见,迄今只报道了60多例。其中,多数(93%)为ABCC8基因复合杂合突变,少数(4%)为KCNJ11基因复合杂合突变。另外,还报道了2例(3%)ABCC8/KCNJ11复合杂合突变。本研究中,9例患儿均携带ABCC8基因复合杂合突变,经遗传学确诊为KATP-HI,符合文献报道的复合杂合突变基因分布特征。
随着研究不断深入,迄今已报道52种ABCC8复合杂合突变,较常见的突变包括p.A640V、p.R168C、p.Q1196*、p.R598*等。报道过的复合杂合突变中,只有1例为新生突变(c.IVS10+1G>T/ p.T1531A),其中p.T1531A为新生突变,其余突变多来自表型正常的父母。已报道的ABCC8基因复合杂合突变类型复杂多样,有错义突变、移码突变、剪切突变和缺失突变等。
本组9例患儿共检测到18个ABCC8基因突变,遗传方式均为常染色体隐性遗传。其中9个突变已有文献报道,另9个为中国人首次报道,尚未见相关数据库收录(参考数据库:Pubmed及ClinVar数据库),其变异均可能有致病性。除p.A640V、p.R275Q突变为新生突变外,其余16个突变均来自临床表型正常的父母,符合文献报道的ABCC8基因复合杂合突变特征。本组病例突变类型共3种,错义突变:p.Y179C、p.W1246R、p.N72K等12个;无义突变:p.R598*、p.Q1196*等4个;移码突变:p.T1042QfsTer75、p.E945RfsTer25。
文献资料显示,携带ABCC8基因复合杂合突变的CHI患者多起病较早,多于新生儿期起病,少数于婴儿期起病。迄今报道资料完善的42例患者中[3-5,7-18],新生儿期起病38例(90.5%),1~6个月起病3例(7.1%),6个月后起病1例(2.4%)。本组患儿中,新生儿期起病6例(66.7%),1~6个月起病2例(22.2%),6个月后起病1 例(11.1%),基本符合文献报道的起病年龄特征。
携带ABCC8基因复合杂合突变的KATP-HI患儿出生体质量多为巨大儿[3-5,7-18]。在既往报道出生体质量明确的33例病例中,低出生体质量儿1例(3.0%),正常出生体质量儿8例(24.2%),巨大儿24例(72.7%)。本研究中,低出生体质量儿1例(11.1%),正常出生体质量儿2例(22.2%),巨大儿6例(66.7%),出生体质量占比与文献报道基本一致。
根据胰腺组织学特征的不同,KATP-HI可分为弥漫型、局灶型和非典型3种类型。既往研究显示,携带ABCC8基因复合杂合突变的CHI患儿组织分型大多数为弥漫型(89%),少数为局灶型(7%),只有1例(4%)为非典型[3-5,7-18]。Zhang等[7]于2015年首次报道1例中国汉族人ABCC8基因复合杂合突变病例,该例术前18F-L-DOPA-PET胰腺扫描显示胰头有局灶性病变,胰腺病理提示为非典型,这也是目前唯一1例非典型的ABCC8基因复合杂合突变病例。本组仅2例行18F-L-DOPA-PET胰腺扫描,结果均为弥漫型。其余7例组织学分型尚不明确,根据既往研究资料推测为弥漫型可能性大。
KATP-HI治疗分为内科治疗和外科治疗。二氮嗪是CHI治疗的一线治疗药物,二氮嗪无效患儿可选用奥曲肽[8]。大多数ABCC8和/或KCNJ11导致的KATP-HI对二氮嗪药物治疗无效,需行不同程度的胰腺切除术[9]。功能缺陷的KATP通道导致质膜持续去极化,胰岛素大量分泌,二氮嗪作为一种KATP通道激动剂,使通道处于开放状态,从而抑制胰岛素分泌,部分突变的复合杂合子之间相互作用,导致细胞内保留通道复合物,故二氮嗪仍能发挥作用[19]。
本研究对既往报道ABCC8基因复合杂合突变的文献进行统计[3-5,7-18],大多数ABCC8基因复合杂合突变导致的CHI对二氮嗪药物治疗无效,27例中有效占15%,无效占85%。本研究中,8例患儿曾应用二氮嗪试验性治疗,有效3例(37.5%),无效5例(62.5%),与文献报道基本一致。文献资料显示,ABCC8基因复合杂合突变导致的7例CHI患儿,应用奥曲肽治疗部分有效,有效占43%,无效占57%。本组4例曾应用奥曲肽治疗,均有效,这种有效性的差异,考虑与本研究的样本量较小有关。既往报道的60多例ABCC8基因复合杂合突变病例中[3-5,7-18],23例曾行手术治疗,仅15例描述了术后情况,其中,术后血糖稳定者2例(13%),术后并发糖尿病需胰岛素治疗者3例(20%),术后仍有反复低血糖发作者10例(67%)。本组病例中,1例经胰腺次全切除术治疗,术后仍有低血糖发作,需长期奥曲肽治疗以维持正常血糖水平。
研究资料显示,ABCC8基因复合杂合突变患儿大多数预后不良,可能出现发育迟缓、语言障碍、癫痫、智力低下等,早产儿持续反复的低血糖发作与18个月大时发生脑瘫、发育迟缓、智力低下的风险增高有关,部分患儿经长期药物治疗及饮食控制,血糖维持平稳[13,20]。本研究中,例9于2岁时起病,以抽搐为首发症状,随后诊断为癫痫,提示低血糖对中枢神经系统的损伤可能在疾病诊断之前就已经发生。例3和例5有语言发育迟缓,其中例3为36+5周早产儿,为低出生体质量儿,生后10 d以抽搐和低血糖症状起病,目前纳食少或禁食6 h情况下出现抽搐发作,语言发育迟缓可能是因为患儿为早产儿,且喂养不当,导致严重反复低血糖发作对神经系统造成不可逆损伤。
部分ABCC8基因复合杂合突变患儿通过保守治疗低血糖症状可得到改善,且预后较好,甚至可自行缓解[10]。本研究中,3例(例7、8、9)自行缓解,自行缓解率为33.3%,其中例7和例8对二氮嗪敏感,分别于1岁和 2岁时自行缓解。例9二氮嗪治疗无效,5岁时低血糖症状自行缓解,但合并癫痫,需长期口服抗癫痫药物治疗。
总之,本研究资料显示,中国KATP-HI儿童的ABCC8基因复合杂合突变类型多样,以错义突变为主。携带ABCC8基因复合杂合突变的患儿多为巨大儿,多于新生儿期发病。该型患儿多对二氮嗪治疗不敏感,部分对奥曲肽治疗有效,部分患儿可获得自行缓解。
猜你喜欢
中国CT和MRI杂志(2022年10期)2022-10-18
保健与生活(2022年9期)2022-05-06
健康之家(2021年6期)2021-09-08
人人健康(2020年4期)2020-05-25
考试周刊(2017年26期)2017-12-12
校园英语·下旬(2017年7期)2017-07-14
家庭医药·快乐养生(2017年5期)2017-05-18
科技视界(2016年27期)2017-03-14
妇女之友(2016年11期)2017-01-20
中学生理科应试(2016年7期)2016-05-14
