
单细胞测序方法研究进展
2022-06-13 11:10:50操利超张核子
生物信息学 2022年2期
操利超,巴 颖,张核子*
(1. 西北大学 生命科学学院,西安 710127;2. 深圳市核子基因科技有限公司,广东 深圳 518071)
1 单细胞测序原理
单细胞测序从单个细胞水平上对DNA或RNA进行扩增和测序,主要包括单细胞分选、核酸提取和文库构建、高通量测序和数据分析等。
单细胞分选是单细胞测序的第一步,如何低成本的获取大批量高质量完整的单细胞对于单细胞测序非常重要。Gross等[1]详细介绍了5种单细胞分选方法,即有限稀释法(Limiting Dilution)、显微操作法(Micromanipulator)、荧光激活细胞分选(Fluorescence activated cell sorting,FACS)、激光显微切割(Laser capture microdissection,LCM)和微流控分选(Microfluidics),这5种单细胞分选方法各有利弊,其中,微流控分选方法由于通量高、成本低等原因而广泛应用于各种商业化单细胞测序平台,例如 10X Genomics公司的Chromium系统就是利用微流控技术进行单细胞分选,通过控制流体流动来实现在微尺度上对目的细胞进行分离。
分离得到单细胞后经过细胞溶解获取DNA或RNA。在传统的高通量测序流程中,提取的DNA或RNA需要进一步纯化后才能应用于扩增,而在单细胞测序中,为了避免DNA或RNA在纯化中丢失,目前大部分流程中去掉这一步骤。后述的文库构建前处理和文库构建流程在不同的测序平台和方法有所不同,但基本上都是基于单分子标签(Unique Molecular Identifier,UMI)和细胞标签(Cell label, CL)的技术,最终形成具有特异标签标记的单细胞文库,文库构建完成后即进行高通量测序和数据分析。在进行数据分析时,单分子标签为每个细胞甚至每个基因或转录本提供特异的识别码,使得精确分析每个细胞的功能和特性成为可能。
2 单细胞测序分类
单细胞测序主要包括单细胞基因组测序(Single cell DNA sequencing, scDNA-seq)、单细胞转录组测序(Single cell RNA sequencing, scRNA-seq)和单细胞表观组测序(Single cell epigenome sequencing),这三种测序类型可以从不同角度揭示细胞各个阶段的功能和特性。
2.1 单细胞基因组测序
单细胞基因组测序可用来分析单细胞水平的点突变和拷贝数变异,用于揭示细胞群体差异、细胞进化关系等,可最真实的获得单克隆癌细胞的具体突变来源及精准的突变频率,以及区分癌症发生、发展、演化过程中的主动与被动突变等。其主要难点是获得高覆盖度高保真性的全基因组扩增产物,因此,在单细胞全基因组技术发展过程中,全基因组扩增技术(Whole-genome amplification,WGA)也经历了几次重大的变革。WGA主要有三种方式,包括简并寡核苷酸引物PCR (Degenerate Oligonucleotide-Primed Polymerase Chain Reaction,DOP-PCR)、多位点置换扩增(Multiple Displacement Amplification,MDA)、 MDA与PCR相结合的方法(Multiple Annealing and Looping-Based Amplification Cycles,MALBAC)等三种。DOP-PCR方法的原理是在引物的3’端含有6bp的随机序列,可以与基因组DNA随机结合,从而实现对全基因组高质量的扩增[2];MDA方法引入了phi29 DNA聚合酶,使其与随机的六聚体发生反应,phi29 DNA聚合酶具有很强的链置换特性,在等温条件下,能够扩增出的50-100 kb的DNA片段[3]。相对于DOP-PCR,MDA的覆盖度和均匀性有了明显提升,但是这种方法并不是无偏倚性、无误差的。鉴于MDA方法扩增得到的基因组覆盖度不均匀,Zong等[4]开发了一种新的基因组DNA扩增方法,即MALBAC。该方法将MDA与常规的PCR结合起来,利用部分碱基简并化的杂交引物与模板退火结合,在链置换酶的作用下进行扩增,扩增中间产物的3’端带有特异性引物标记,经过一轮扩增后,互补的标记位于5’端,两端的标记互补成环,成环后的扩增子通过常规PCR进行扩增。
2.2 单细胞转录组测序
单细胞转录组测序可对单细胞中mRNA进行基因表达定量、功能富集、代谢通路等分析,可以解决传统转录组测序技术在早期胚胎发育、干细胞、癌症、免疫等研究领域中存在的样品量极低或细胞异质性的问题,是在单细胞水平研究基因表达强有力的工具。单细胞转录组测序需要对获取的RNA进行逆转录,收集cRNA产物并扩增测序。各种成熟的单细胞转录组测序方法的标准操作规程(Standard Operating Procedure, SOP)在2013年前后均有文章发表(见表1)。单细胞转录组测序方法很多,在实际的项目中,需要根据实际情况综合评估,选择合适的测序方法。Christoph等[5]利用6种不同的单细胞转录组测序方法对583个小鼠胚胎干细胞进行分析。相对于Smart-seq和Smart-seq2,其他检测方法CEL-seq2、Drop-seq、MARS-seq和SCRB-seq采用了单分子标签技术,这可以提高测序的准确性,而Smart-seq和Smart-seq2在建库时能获取全长转录本,这有利于检测到更多的基因,经过标准化的分析流程处理后,文章对这6种测序方法进行了全面的对比,包括测序质量、测序深度、检测到的基因数、有效的细胞数、准确性和灵敏度等。通过综合考虑分析,给出的结论是当对大量细胞进行转录组定量分析时,采用Drop-seq方法性价比最高,而对于少量细胞,MARS-seq和SCRB-seq和Smart-seq2方法更有效。
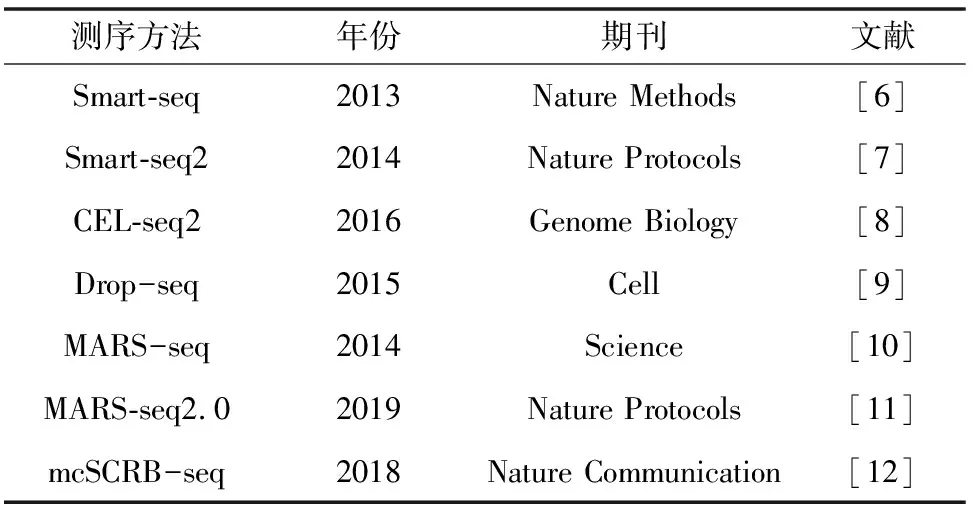
表1 不同单细胞测序方法学统计表Table 1 Different single cell sequencing methodologies
2.3 单细胞表观组测序
单细胞表观组测序可从单细胞水平获得全基因组范围内的甲基化水平数据,对于表观遗传学的时空特异性研究具有重要意义。单细胞表观基因组测序主要是结合单细胞基因组测序和传统的表观组高通量测序方法(如BS-seq和ChIP-seq等)。如Guo等[13]利用单细胞全基因组简化胞甲基化测序(Single cell reduced representation bisulfite sequencing, scRRBS)的方法对小鼠胚胎干细胞进行测序,可在单个细胞中检测到高达150万个CpG位点的甲基化状态,同时,该技术可以检测单倍体精子细胞中单个CpG位点的甲基化状态,可探索单个细胞DNA甲基化的动态变化。RRBS方法只能检测到全基因组上5%左右的CpG位点的甲基化信息,且这些位点主要集中在CpG相对密集的区域,比如CpG岛、启动子等,但是在胚胎早期发育过程中,全基因组范围内的甲基化状态是变化的,为了检测到更全面的甲基化信息,Zhu等[14]利用重亚硫酸盐处理后接头标记技术(Post-bisulfite adaptor tagging,PBAT)对人植入前胚胎发育的各个阶段进行了深度测序,平均每个细胞能捕获全基因组上的20%的CpG位点,分析发现数以万计的基因组位点表现出从头开始的DNA甲基化(De novo DNA methylation)。这一发现表明,全基因DNA甲基化重编程过程在植入前胚胎发育过程中甲基化和去甲基化是处于动态平衡的。同时,通过DNA甲基化分析,可以追溯早期胚细胞的遗传谱系,为破译早期人类胚胎中DNA甲基化重新编程的秘密铺平了道路。基于染色质免疫共沉淀技术(Chromatin Immunoprecipitation,ChIP)的测序方法,Rotem等[15]结合微流控和DNA特征码技术进行了数千个单细胞测序,从单细胞水平收集染色质数据,进一步分析了表观遗传各方面的异质性,而这在转录水平是无法获取的。
2.4 单细胞多组学测序
此外,单细胞多组学研究(Single cell multi-omics sequencing)也逐步在科学研究中得到应用。如Angermueller等[16]对61个小鼠胚胎干细胞同时进行单细胞转录组和表观组测序,分析了DNA甲基化异质性和转录水平异质性的关联。Macaulay等[17]利用基因组和转录组平行测序(Genome and Transcriptome Sequencing, G&T-seq)方法对来源于小鼠和人类的220多个单细胞进行测序,通过关联分析发现了以前无法单独从DNA或RNA测序推断出的细胞特性。
3 单细胞测序技术平台
3.1 单细胞分离和标记平台
单细胞测序技术一般是结合单细胞分离技术和特异性分子标签技术将单个细胞标记上特定的标签,然后进行高通量测序的技术。目前,国内外大规模单细胞技术使用的单细胞分离和标记平台主要有基于10X Next GEM技术的ChromiumTM系统、BD RhapsodyTM单细胞分析系统、Illumina©Bio-Rad©单细胞测序解决方案、ICELL8 单细胞系统、C1TM单细胞全自动制备系统等。各个单细胞分离和标记平台的原理、特点及应用(见表2)。在实际的项目中,需要结合项目的特点,考虑多种因素,选择一种最能满足实验且性价比高的平台。

表2 不同单细胞分离和标记平台的原理、特点及应用Table 2 Principle, characteristics, and application of different single cell separation and labeling platforms
3.2 单细胞高通量测序平台
单细胞测序技术使用的高通量测序平台有很多,如illumina系列、BGISEQ系列、Roche 454、ABI solid、Ion Proton等。目前,应用于单细胞测序的高通量测序平台主要是illumina系列,但其他测序平台也逐渐被证明可应用单细胞测序。Chen等[18]在illumina Hiseq2000和Ion Proton平台上分别利用低覆盖度的单细胞全基因组测序的方法对相同的样本进行测序,并从可重复性、测序错误率、一致性、灵敏度和特异性等方面进行比较分析,结果表明,两种平台各有优缺点。
最近,有研究表明,BGISEQ测序平台也可应用于单细胞测序。Natarajan等[19]选取了468个单细胞对应1,297个cDNA样本,分别采用SMARTer和Smart-seq2的方法在BGISEQ-500和Illumina HiSeq平台上进行单细胞测序,本文首次从灵敏度和准确性上比较在两种平台在单细胞转录组测序上的应用,结果表明,BGISEQ-500可完成高质量低成本的单细胞转录组测序。
4 单细胞测序数据分析
单细胞测序分析中,对于不同的测序类型和研究目的,会采用不同的分析流程。对于单细胞基因组测序和单细胞表观组测序而言,数据分析流程与传统的高通量测序数据分析方法类似[13, 20]。在单细胞转录组测序数据分析中,常见的分析内容包括基因表达(gene expression)、可变剪切(alternative splicing)、T细胞受体谱 (T cell receptor profiling)或B细胞受体谱 (B cell receptor profiling)、细胞聚类(Cells clustering)、拟时序分析(Trajectory analysis)等,常见的单细胞转录组测序数据分析流程(见图1),一些主流的分析软件总结(见表3)。

表3 单细胞转录组测序分析软件Table 3 Single cell transcriptome sequencing analysis toolkits
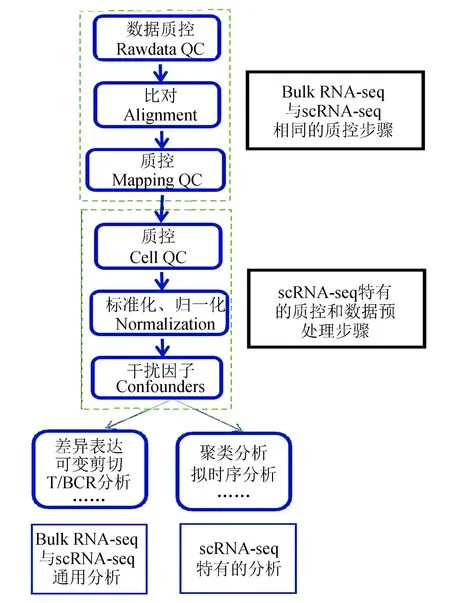
图1 单细胞转录组测序分析流程Fig.1 Analysis workflow of single cell transcriptome sequencing
在单细胞转录组测序数据分析中,细胞聚类和拟时序分析是单细胞测序特有的分析内容,下面将重点阐述。细胞聚类是单细胞测序数据进一步分析的基础。细胞聚类的基本原理是根据细胞的特征(比如基因表达等)将大量的细胞,通过聚类算法将细胞分成不同的亚群的过程。Dai H等[21]通过构建单细胞的network,将network降维到二维的矩阵,并代替原有的不稳定的基因表达矩阵,用以细胞聚类和拟时序分析,同时,该种方法可以发现一些有意义的在传统差异基因表达分析中漏掉的认为不重要的基因。Elham等[22]利用Drop-seq方法对45 000个免疫细胞进行单细胞测序,通过自己开发的分析流程,通过贝叶斯聚类和标准化方法,提出差异表达分析不能完全体现亚群间差异,得到非常稳定的协方差模式(Covariance pattern)不同的亚群。Xie等[23]用有监督的机器学习方法大大提高了细胞聚类的效率, 在单核2.3 GHz的个人电脑上,训练19万个细胞的训练集只需要5分钟,而做1万个细胞的分类不到1分钟,而常用的聚类软件Seurat,在对1W个细胞进行聚类时,时长需要以小时来计算,内存用量约15G-20 G,分析结果表明在准确性上还有待提升,这主要跟提供的数据训练集有关。同样,Feiyang Ma等[24]利用神经网络模型对小鼠细胞和人源免疫细胞数据集进行训练,利用得到的训练结果来预测小鼠白细胞、人源外周血单核细胞和人源T细胞亚型,分析结果快速而准确,表明该方法可以用来优化目前的单细胞测序分析流程。随着单细胞测序样本量的增大,传统的聚类分析方法需要耗费大量的资源,机器学习的引入会是一个很有潜力的解决方向,但需要进一步提高聚类的准确性和扩展应用场景。
拟时序分析是基于大量单细胞的基因组学数据,通过生物信息学算法来推断这些细胞的发育时序。拟时序分析对于研究某一特定细胞类型的转化,如CD8+T细胞的激活和耗竭、M1/M2型巨噬细胞极化等,往往具有一定的生物学意义。进行拟时序分析的软件有很多,Saelens等[25]针对110个真实数据集和229个模拟数据集,利用45种拟时序分析工具进行分析和对比,分析结果表明工具和方法的选择,主要取决于数据集维度和拓扑结构。
5 单细胞测序研究热度分析
单细胞测序是为了解决传统高通量测序的局限性,比如无法获取单个细胞特有的遗传信息,可能会丢失低丰度的信息,无法检测难以培养富集的微生物等。因此,单细胞测序具有传统高通量测序无法比拟的优势。2011年,《自然方法》杂志(Nature Methods)将单细胞测序列为年度值得期待的技术之一,2013年,《科学》杂志(Science)将单细胞测序列为年度最值得关注的六大领域榜首,2018年,《科学》杂志(Science)的年度十大科学突破之一就是单细胞测序的重要一环“单个细胞分离并逐个测序(Development cell by cell)”的研究进展。由此可见,单细胞测序作为一种技术手段被广大科研工作者寄予厚望。
以"single cell sequencing"[All Fields]为关键词去NCBI-pubmed数据库搜索,统计搜索到的文章数(见图2),可以发现从2011年到2020年间,单细胞测序发表的文章数基本上呈指数增长(截止至2020年10月16日)。
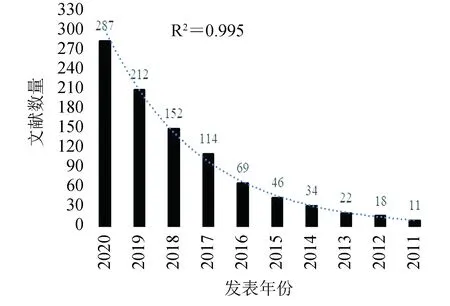
图2 以“单细胞测序技术”为关键词在NCBI-pubmed库中的搜索情况(截止至2020年10月16日)Fig.2 Statistics of published literature in NCBI-pubmed database using “single cell sequencing” as keyword (by October 16, 2020)
在科学网基金页面(http://fund.sciencenet.cn/),以“单细胞测序”为项目名称关键词搜索,查询结果有1 218项,累计金额为75 772万元,项目涉及学科分类广,主要集中在生命科学和医学科学领域,分布图(见图3)(查询结果截止至2020年10月16日)。

图3 以“单细胞测序”为关键词在科学网基金页面查询得到的项目分布统计图(截止至2020年10月16日)Fig.3 Statistics distribution of projects on the Science Net using “single cell sequencing” as keyword (by October 16, 2020)
在美国国立卫生研究院(NIH)官网上(https://projectreporter.nih.gov/),以“single cell sequencing”为关键词搜索,查询结果按照经费申请机构进行统计,我们通过经费申请机构可以简单了解其研究方向或研究领域,最终的统计结果(见图4)(查询时间跨度为2018年至2019年)。从图中可以看出,在单细胞测序研究中,肿瘤和常见疾病相关研究机构申请到的项目或研究经费较多,由此可以粗略推断单细胞测序的热点研究领域。
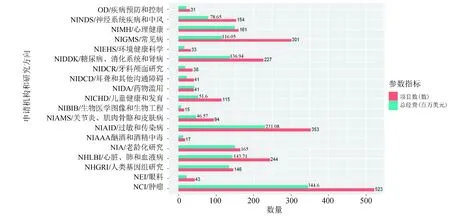
图4 以“single cell sequencing ”为关键词在美国NIH页面查询得到的项目分布统计图(2018-2019年)Fig.4 Statistics distribution of projects on the NIH website using “single cell sequencing” as keyword (2018 - 2019)
由此可见,单细胞测序在近年来一直是科研界的研究热点,被广泛应用于各个领域。
6 单细胞测序的应用
单细胞测序在不同研究领域得到应用,比如干细胞和发育生物学、肿瘤、免疫学等领域。
发育生物学作为生命基本过程的基础学科,传统的高通量测序无法详细和系统的研究所有器官各种细胞的分子状态和分化途径。而单细胞测序可以分离单个细胞,对其中的遗传信息进行分析,构建高分辨率的不同组织发育阶段的基因表达谱。Takahashi K等[34]研究发现,已分化的成纤维细胞可以在四种反转录因子OCT3/4, SOX2, c-Myc 和 KLF4的作用下重分化成多能干细胞,这一发现有望在多种疾病的机制研究和治疗中发挥重要的作用。Brunskill EW等[35]利用单细胞测序研究了肾脏器官发育过程中的基因表达谱,发现在不同的发育阶段,结构相同的细胞群却有着不同的来源。
在肿瘤研究中,了解肿瘤细胞内异质性对肿瘤的发生发展、其耐药性的影响和重新定义细胞亚型等非常重要。传统的高通量测序方法只能针对大量细胞群体进行研究,只是细胞群体的总平均反应,无法反应每个细胞的真实情况。而单细胞测序能够揭示单个细胞的基因结构和基因表达状态,反应细胞间的异质性。Li H等[36]利用单细胞测序技术对11个结直肠癌细胞和对应的正常粘膜细胞进行测序,利用参考成分分析(reference component analysis,RCA)算法进行聚类分析鉴别出两种不同的纤维母细胞瘤亚型,同时,在其中发现了一些与上皮间充质干细胞分化相关的基因表达上调,为肿瘤细胞异质性表征提供了一个很好的方法。Tirosh等[37]从19个黑色素瘤患者中分离了4 645个细胞进行单细胞测序,研究发现,非恶性肿瘤细胞会根据细胞类型如T细胞、B细胞、巨噬细胞、血管内皮细胞等聚类,而不同病人来源的恶性肿瘤细胞会被分成不同的聚类,表明肿瘤细胞异质性的存在。Baslan等[38]综述了单细胞测序技术在研究肿瘤异质性和肿瘤细胞演化中的作用,并以肺癌为例说明了单细胞测序的应用场景。
在免疫学领域,由于传统的免疫学分析方法取样来自于大量细胞,导致分析结果低估了单个免疫细胞的多样性,单细胞测序可以更加精确地检测单个免疫细胞的遗传物质,从而理解机体复杂的免疫机制。Villani等[39]通过对来源于健康献血者的2 400个细胞进行单细胞测序,发现了6个DC细胞和4个单核细胞亚型,该研究是对pDCs分类的重新鉴定和修订,这一研究成果将使得对疾病和健康的免疫检测更加精确。
7 挑战和展望
相对于传统的高通量测序,单细胞测序检测的技术难点不在于测序本身,而在于单个细胞中核酸物质太少,以至于难以直接检测。因此,单细胞测序的关键技术之一时需要对单细胞中极微量的DNA进行高质量、大幅度的扩增,目前已经有一些相对成熟的扩增方法,但是这些扩增技术都存在一些局限性,比如扩增区域不全而导致未扩增的区域无法被测序,而且扩增会存在偏向性(Bias),导致扩增不均一,这些问题都会给后续的生物信息分析带来很大的挑战。同时,FFPE样本的实验处理和数据分析优化很重要[38],因为临床上有很多样本是FFPE样本,而这些样本很容易降解,从而使其进行单细胞测序存在一些挑战。除此之外,对于大量样本量的单细胞测序来讲,会产生海量的测序数据,这对于数据的存储、分析带来了挑战。
近年来,云平台在高通量测序领域得到广泛的应用,而数学、物理学等学科为生物信息学的发展提供了基础算法,这使得生命科学大数据的计算、存储和应用成本大大降低。同时,单细胞测序成本的降低,使其广泛应用于生命科学各个领域,这也积累了海量的单细胞测序数据,而缺少高效精准的单细胞测序数据分析方法或工具阻碍了单细胞测序的进一步发展。目前,结合人工智能和生物信息算法,已经在单细胞测序数据分析领域取得了突破。如Xiong等[40]利用人工智能深度学习算法,结合变分自编码器和高斯混合模型,提取单细胞ATAC-seq(Assay for Transposase-Accessible Chromatin using sequencing)数据的隐层特征,将问题从复杂稀疏的高纬度的染色质开放图谱空间投射到了简单抽象的低纬度特征空间,这种处理不但可以发现与解析细胞特异性的染色质图谱模式,还通过相似细胞信息共享,填补了技术限制导致的缺失值,从而巧妙地解决了单细胞ATAC-seq数据中高维度、稀疏性、二值化等问题。而Cheng等[41]开发的基于机器学习的分析管道潜在细胞分析法(Latent Cellular Analysis,LCA),通过将隐元胞状态的余弦相似度度量与基于图的聚类算法相结合,为种群数量推断、降维、特征选择和技术变异控制提供启发式解决方案,且无需显式基因过滤。Xie等[42]对正常肺中6种间充质类型细胞和纤维化肺中7种间充质类型细胞进行单细胞RNA测序,并通过机器学习的方法,确定了它们的分化轨迹,从而为了解成纤维细胞的结构和成纤维细胞在纤维化疾病中的作用提供了新的资源。Duan等[43]基于人工智能主题模型(Topic Modeling)的计算框架MUSIC(Model-based Understanding of Single cell CRISPR screening),用以有效地对单细胞CRISPR筛选数据进行分析,用以揭示数据本身所体现的生物学意义。He等[44]基于半监督学习的单细胞测序数据填补方法DISC ( Deep learning Imputation model with semi-supervised learning for Single Cell tranomes ),该方法可以利用少量的表达出来的基因信息及数据庞大的缺失表达基因之间的表达结构,可以实现基因表达分布修复、差异基因预测、基因表达相关性预测和稀有细胞类型预测等,为单细胞测序数据分析提供了重要的技术支撑。
8 结 论
相较传统的混合细胞测序,单细胞测序技术能够在复杂的群体组合(如肿瘤)中对不同细胞类型进行区分,为了解各种发育、生理和疾病系统背后的过程提供了一个强大的方法,这也使得单细胞测序成为科研界的一个研究热点。大量关注和资金的流入使得单细胞测序技术飞速发展,各种测序平台和数据分析算法层出不穷。然而由于单细胞测序数据量大的特点,使得数据分析的难度高,精度差成为阻碍该技术发展的主要因素。但随着人工智能的发展,越来越多的研究者将人工智能技术用于单细胞测序数据的分析,并取得了不错的成果。相信在未来,人工智能作为大数据分析的一个有效的解决方法而被广泛应用于单细胞测序数据分析领域。
猜你喜欢
国际太空(2023年1期)2023-02-27 09:03:42
科学(2020年4期)2020-11-26 08:27:16
透析与人工器官(2020年1期)2020-11-16 01:42:34
铁道通信信号(2019年8期)2019-10-10 05:06:00
中国发展观察(2017年8期)2017-04-26 03:51:50
生殖医学杂志(2015年11期)2015-02-28 16:32:12
化学工业与工程(2015年1期)2015-02-10 03:01:41
现代检验医学杂志(2015年2期)2015-02-06 02:00:48
沈阳医学院学报(2014年4期)2014-12-27 13:44:30
遗传(2014年3期)2014-02-28 20:58:49
