
HPRT1 基因变异致Lesch-Nyhan 综合征1 例报告
2022-06-09 05:55李逢潮章印红朱宝生韩思琪蔡世岩
临床儿科杂志 2022年6期
李逢潮 章印红 吕 涛 朱宝生 韩思琪 蔡世岩 李 利,4
1.昆明理工大学医学院(云南昆明 650500);2.云南省第一人民医院儿科(云南昆明 650032);3.云南省第一人民医院医学遗传科(云南昆明 650032);4.昆明理工大学附属医院(云南昆明 650500)
Lesch-Nyhan 综合征(Lesch-Nyhan syndrome,LNS,OMIM:300322)是一种罕见的X 连锁隐性遗传病,国外一项为期20 年的回顾性研究报道,平均发病率约为1/55 万[1],是由于次黄嘌呤-鸟嘌呤磷酸核糖转移酶(hypoxanthine-guanine phosphoribosyl transferase,HGPRT)缺乏所致,临床表现为运动障碍(肌张力障碍、舞蹈病和痉挛)、智力障碍、高尿酸血症及其后遗症(痛风、肾结石、肾功能衰竭)和自残行为[2]。该病起病早,在自伤行为出现前,易被误诊为脑瘫,延误诊治,随着基因检测技术的迅速发展,国外文献中LNS 的研究报道已不罕见,但国内确诊为本病且完善基因检测的病例仍较少,故本文对1 例明确诊断的LNS 患儿进行介绍。
1 临床资料
患儿,男,8岁7个月,汉族,因发育落后8年余,肌张力异常,于2020年7月27日至云南省第一人民医院门诊就诊。患儿4月龄时发现不会抬头、双手握拳;9 月龄时在外院诊断为“脑瘫”;康复治疗2 年,效果不佳,后间断在外院诊断、治疗(具体不详)。现8岁7个月,不能独坐,不会站,不会拿东西,不会讲话,会哭、会笑,只能用情绪表达喜怒哀乐,能听懂简单指令,认识爸爸、妈妈,生活不能自理。
患儿系G1P1,孕39周,顺产,出生体质量3 500 g,否认围生期缺氧窒息史,生后出现黄疸,短时消退,人工喂养,生后无喂养困难,母妊娠期无疾病史。父母体健,非近亲婚配,否认家族性遗传病史。
体格检查:身高119cm(<-2 SD),体质量20 kg(<-2SD),头围50.5 cm(-0.67 SD)。一般情况尚可,易激惹,无特殊面容,双侧耳廓可见黄白色米粒大小结节,无伸舌、流涎。心、肺、腹(-)。双手呈握拳状,不能独坐,扶腋下可站立,呈尖足。四肢肌张力高,双侧膝腱反射(+++),双侧巴氏征(+)。
辅助检查:6 月龄时Gesell 评分,大运动41,精细动作16,认知水平50,语言75,社交-行为40 ;9月龄听力诱发电位、双髋关节X 线片均正常;1 岁2 个月颅脑MRI 正常;1 岁3 个月外周血染色体、血尿串联质谱未见明显异常。8 岁7 个月血生化示尿酸913 μmol/L(178~416 μmol/L),3 个月后复查871 μmol/L,余正常;腹部B 超示双肾髓质回声增强,其内多发点状强回声声像(考虑双肾痛风肾);血常规、尿常规、甲功、血氨、血乳酸、血串联质谱、高通量测序CNVs(Copy number variants)变异检测等未见明显异常。
因患儿病程长,不明原因发育迟缓伴肌张力高,且血尿酸明显偏高,高度怀疑遗传代谢性疾病,经医院伦理委员会批准(No.KHLL 2021-KY 098)及患儿父母知情同意后,采集患儿及其父母外周血各2 mL(EDTA 抗凝),从受检者外周血中提取基因组DNA,构建基因组文库,捕获相关目的基因片段并进行富集;富集的目的基因片段利用Illumina HiSeq高通量测序仪进行测序。测序数据运用NextGene V 2.3.4 软件与人类基因组hg 19 参考序列进行比对,并对目标区域的覆盖度和测序质量进行评估;依据严格的筛选标准对变异进行过滤,并添加人类基因变异数据库(Human Gene Mutation Database,HGMD,http://www.hgmd.org/)和蛋白功能预测软件等的相关注释信息。根据HGMD查找变异位点的收录情况,参考美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)基因变异解读指南[3]从数十万个变异中筛选与疾病明确相关或可能与疾病相关的变异,并对变异位点进行综合评价。对明确或可能与受检者临床表型相关的基因变异采用Sanger 测序进行验证。在X染色体HPRT1基因3号外显子上检测到1个相关变异(NM_000194.2:c.200_201 delTG),该变异为移码变异,可导致蛋白质截短,为半合子变异,经ACMG评级为疑似致病性变异,经Sanger测序验证发现该变异遗传自患儿母亲,母亲为杂合携带(图1)。在千人基因组、ExAC和gnomAD外显子数据库中均未发现此变异(PM2,pathogenic moderate)。
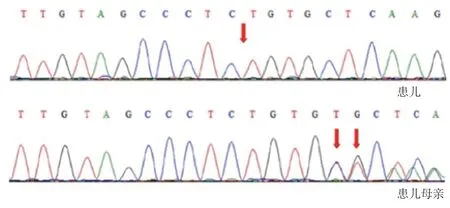
图1 患儿及其母亲HPRT1 基因变异Sanger 测序图
该患儿确诊为Lesch-Nyhan综合征,建议低嘌呤饮食,适当多饮水,行HLA-B*5801 基因检测,结果为阴性后,予别口服嘌醇片0.1 g,5 mg·kg-1·d-1,qd,碳酸氢钠片0.5 g,qd。对患儿进行随访,口服别嘌醇片6 个月后,尿酸水平下降至437 μmol/L,仍无自伤行为(具体诊断、治疗、随访时间轴见图2)。
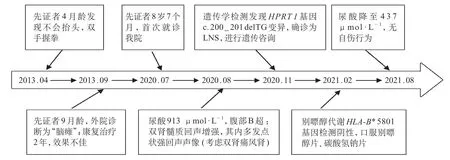
图2 先证者诊断、治疗、随访时间轴
2 讨论
Lesch-Nyhan综合征是由于HPRT1基因变异所致,该基因位于Xq26.2-q26.3,由1 395 bp组成,目前全球已报道的变异达600 余种,占比最高的是错义变异,其次是缺失[4]。Fu 等[5]对2013年前报道的LNS病例进行统计,发现基因型与表型具有相关性,提出编码区的缺失变异、重复、无义变异常常导致Lesch-Nyhan病,变异型中比较常见的是错义变异及剪接变异。
该病临床表现多样,临床上根据HGPRT酶活性分为3型,①经典型(Lesch-Nyhan病,LND):HGPRT酶活性<1.5%,临床表现高尿酸血症、神经系统异常,多伴有自伤行为;②HGPRT相关的神经功能障碍(HGprt-related neurological dysfunction,HND) :HGPRT酶活性为1.5%~8.0%,临床表现为高尿酸血症及神经系统异常,但神经系统受累较轻,多无自伤症状;③HGPRT 相关高尿酸血症(HGprt-related hyperuricemia,HRH):HGPRT 酶活性>8.0%,临床仅表现为高尿酸血症;后两型也称为LND 的变异型[6]。
LNS患儿在产前和新生儿期大多是正常的,3~6个月时出现坐、爬等运动发育迟缓症状;8~12个月逐渐出现锥体系及椎体外系症状,并且常被误诊为脑瘫,大多数患儿有轻度或中度的智力缺陷,但严重的智力障碍是比较罕见的[7-8];经典型LNS患儿1~3岁开始出现自残行为,也可延迟至十几岁出现。该病最常见的症状是高尿酸血症,并且随着年龄的增大,在青春期出现尿路结石、痛风等症状,并逐渐演变为肾功能衰竭[9]。另外也可有小头畸形、吸入性肺炎、肺栓塞、气管憩室、巨细胞贫血、髋关节脱位、癫痫发作等[10-11]。典型的LNS患儿在自伤行为出现后易被诊断,但由于变异型缺乏或只有非常轻微的行为和神经表现,所以常常以肾结石或痛风作为最初的临床表现就诊[12],需完善血尿酸、尿尿酸、泌尿系统彩超等检查筛查,尽快完善HPRT 1基因确诊,有条件者可完善HGPRT酶活性测定检测判断预后。
本例患儿出生时无明显异常,4月龄时发现发育落后,曾被诊断为脑瘫,进行康复治疗,本次就诊血生化提示血尿酸明显升高,有肾结石,且双侧耳廓可见痛风石,但目前尚无关节损伤及痛风症状,完善基因检测,发现在X染色体HPRT1基因3号外显子上存在 c.200_201delTG,确诊为LNS。
目前对于LNS患者的神经和行为障碍机制尚不清楚,无特异性治疗,只能对症治疗。针对高尿酸血症,首选别嘌呤醇降尿酸治疗,但为预防别嘌醇超敏反应,在使用前需进行HLA-B*5801基因检测,结果为阴性者方可使用[13];也可采用低嘌呤饮食,增加饮水量,加用柠檬酸钾、碳酸氢钠片促进尿酸排泄,减少高尿酸血症所致并发症,但降尿酸治疗并不能减少神经精神症状。Jacomelli等[14]用嘌呤核苷磷酸化酶(purine nucleoside phosphorylase,PNP) 抑制剂阻断次黄嘌呤上游的产生来治疗LND 患者的尿酸、次黄嘌呤和黄嘌呤过量,这为LNS 高尿酸血症提供了一种新的治疗策略,并预防了黄嘌呤结石的产生。针对肌张力障碍及痉挛者可选用苯二氮卓类药物、γ-氨基丁酸抑制剂等。意大利一项研究证明,鞘内给巴氯芬对控制肌张力障碍有效,并可部分改善自伤行为[15]。针对自伤行为,常常采用人为约束、行为及心理治疗,保护牙齿、口唇及手指等,必要时拔除牙齿。有研究证明多巴胺能药物、S腺苷甲硫氨酸、深部脑刺激(deep brain stimulation,DBS)、肉毒杆菌毒素注射等可以减少LNS 患者的自伤行为[16-19]。LNS患者通常因喂养困难导致缺铁性贫血,使平均红细胞体积正常化,从而掩盖了巨红细胞贫血,故通常全血细胞检测不能发现,需常规行铁含量的测定,巨细胞贫血是该病的一个特征,在LNS 的早期评估中具有重要的作用,不需口服叶酸及维生素B12治疗[10]。LNS患者通常能活到20~30岁,死亡原因包括肺炎以及肾功能衰竭等。
本例患儿确诊后行HLA-B*5801 基因检测,结果为阴性后,予别嘌醇片及碳酸氢钠片降尿酸治疗,治疗6个月后尿酸水平下降至437 μmol/L。因我院目前尚不能行HGPRT酶活性检测,故患儿未完善酶活性检测,无法进行明确分型,但根据患儿临床表现考虑患儿为HND可能性大,因自伤行为与年龄有相关性,需继续随访患儿有无自伤症状。
综上所述,LNS为一种罕见病,在自伤行为出现前常常被误诊为脑瘫,且康复治疗效果差,故临床上遇到不明原因脑瘫表现者,应常规行血生化检查进行筛查,如合并尿酸增高,建议行基因检测,及早明确病因、对症治疗,做好遗传咨询,必要时行产前基因检测预防该病的发生。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
浙江临床医学(2022年8期)2022-09-14
中国医药科学(2022年1期)2022-05-05
健康体检与管理(2022年2期)2022-04-15
保健与生活(2020年19期)2020-10-26
保健与生活(2020年9期)2020-05-28
特别健康·下半月(2019年9期)2019-09-24
中国保健营养(2019年4期)2019-09-10
家庭医药·快乐养生(2017年6期)2017-06-16
人生十六七(2016年14期)2016-12-01
