
性早熟女童肠道菌群和抗生素耐药性的宏基因组分析
2022-06-08 08:39尹明月董治亚陆文丽王歆琼王俊祺
诊断学(理论与实践) 2022年1期
许 飞,尹明月,王 伟,董治亚,陆文丽,余 熠,王歆琼,王俊祺,肖 园
(1.上海交通大学医学院附属瑞金医院儿科,上海 200025;2.上海市普陀区利群医院儿科,上海 200333)
性早熟是指男孩在9 岁前、女孩在8 岁前呈现第二性征,是最常见的青春发育异常,而女孩发病率为男孩的5~10 倍。中枢性性早熟 (central precocious puberty,CPP) 是由于下丘脑-垂体-性腺轴过早激活引起的;而单纯性乳房早发育(premature thelarche,PT)则是一种不完全性性早熟,表现为女孩8 岁前出现的乳房发育,但无其他第二性征发育。研究发现,全球女孩乳房发育的年龄每10 年就下降约0.24 岁,亚洲更加显著[1],CPP 的发病率为0.01%~0.02%,PT 的发病率为1%~4.7%,且呈逐年上升趋势。遗传、营养、环境和社会经济等一系列因素都对青春发育时间有着重要影响。既往研究提示,肠道菌群在肥胖的发生和发展过程中起着重要作用[2]。同时,肥胖人群具有一定程度的菌群失调[3],而性早熟与儿童肥胖间也存在一定的相关性[4]。因此推测,在儿童性早熟和肠道菌群之间可能存在着一定的联系。
抗生素在医疗事业和畜禽养殖业的大量使用及其在生物体内的不完全代谢,使其在环境中残留,还可能诱导形成抗生素耐药菌和耐药基因[5]。我国是抗生素生产和使用的大国,也是耐药基因扩散的热区,而耐药基因是一种新型污染物[6]。研究表明,儿童时期的 抗生素暴露与其肥胖发生风险呈正相关,在停用抗生素后,肠道菌群虽然可以逐渐恢复,但抗生素暴露对代谢的影响依然存在[7]。儿童肥胖与畜禽用抗生素暴露之间存在明显联系,对儿童生长、发育有着潜在的危害[8]。有关性早熟患儿抗生素暴露的风险目前国内外尚无报道。
宏基因组测序技术是指提取全部微生物基因组的总和而进行检测。相较于传统的16S rRNA 测序方法,该方法不仅可以获得肠道菌群的组成,同时也能获得功能基因的组成,可了解肠道菌群中以前未知或未培养出的重要微生物。本研究纳入女性CPP 及PT 患儿,并设置年龄匹配的健康对照(normal control,NC),以粪便为研究样本,利用鸟枪法宏基因组测序技术对CPP 组、PT 组及NC 组3 组女童的肠道菌群进行特征性分析,旨在分析性早熟及健康女童肠道菌群和耐药基因的特征,为探索抗性基因对儿童生长、发育的影响提供参考。
资料与方法
一、资料
本研究于2020 年6 月至2021 年1 月在我院就诊的女童中进行筛选,纳入并留取粪便样本共40 例。由儿童内分泌专科医师收集研究对象的病史、体格检查、实验室及影像学检查等相关信息。参照2015 年《中枢性性早熟诊断与治疗共识》排除病理性性早熟5 例(MRI 异常2 例,甲状腺功能异常2 例,McCune-Albright 综合征1 例),小于6 岁3 例,阴道异物1 例,最终共31 例采用宏基因组测序,其中去除样本杂质建库失败的2 例。最终本研究纳入21 例,性早熟女童平均年龄为(7.6±0.9)岁,其中CPP 患儿11 例,PT 患儿10 例,同时纳入8 名年龄匹配[平均年龄为(7.2±0.5)岁]的健康女童。
本研究经上海交通大学医学院附属瑞金医院伦理审查委员会批准,研究之前已获得所有受试者及其监护人知情同意。
二、方法
1.粪便样本收集:受试者于家中或医院采集粪便样本。收集管收集粪便样本10 g,平均分为2 份,1 份用于验证实验。标本在收集后15 min 内于-20 ℃保存,收集24 h 内用干冰转移到-80 ℃冰箱中储藏。
2.DNA 样品检测及建库:采用琼脂糖凝胶电泳分析DNA 的纯度和完整性,Qubit 对DNA 浓度进行精确定量。检测合格的DNA 样品用Covaris 超声波破碎仪随机打断成长度约为350 bp 的片段,经末端修复、加A 尾、加测序接头、纯化、PCR 扩增等步骤完成整个文库制备。
3.宏基因组测序:所有粪便样本均送至北京诺禾致源科技股份有限公司进行宏基因组测序,库检合格后使用Illumina Novaseq6000 高通量测序仪进行测序。
4.统计学分析和生物信息学分析:采用R 软件(version 4.0.5)进行统计学分析。涉及的数据若符合正态分布,用均数±标准差来表示,采用ANOVA检验分析;非正态分布使用中间数和四分位数表示,采用Kruskal Wallis 秩和检验。菌群测序数据使用NCBI 数据库进行注解,统计每个样本中的肠道菌群在门、纲、目、科、属、种6 个分类水平的组成。使用主成分分析 (principal component analysis,PCA)和无度量多维标定法 (non-metric multidimensional scaling,NMDS) 分析各样本间物种组成,根据Anosim 分析计算P 值;利用Metastats 方法校正P值,根据Q 值筛选差异性物种[9]。所有统计均为双尾检验,P<0.05 提示差异有统计学意义。
5.抗性基因注释:通过耐药基因数据库(Comprehensive Antibiotic Resistance Database,CARD)对耐药基因进行鉴定挖掘,其核心是抗生素耐药性本体 (antibiotic resistance ontology,ARO),ARO 包含了与耐药基因及其抗性机制等。该数据库收集有关抗生素抗性基因、蛋白质和表型的参考信息,提供了与抗生素耐药性的分子基础有关的数据、模型和算法[10]。本研究采用DIAMOND 软件,将非冗余蛋白序列与该数据库比对,获得基因对应的抗生素抗性功能注释信息,根据功能对应的基因丰度总和计算耐药基因的丰度[11]。
结 果
一、CPP 组、PT 组以及NC 组的基线数据特征
所有研究对象的基线资料见表1,CPP 组、PT组及NC 组间的体质量指数(body mass index,BMI)差异无统计学意义(P=0.687)。
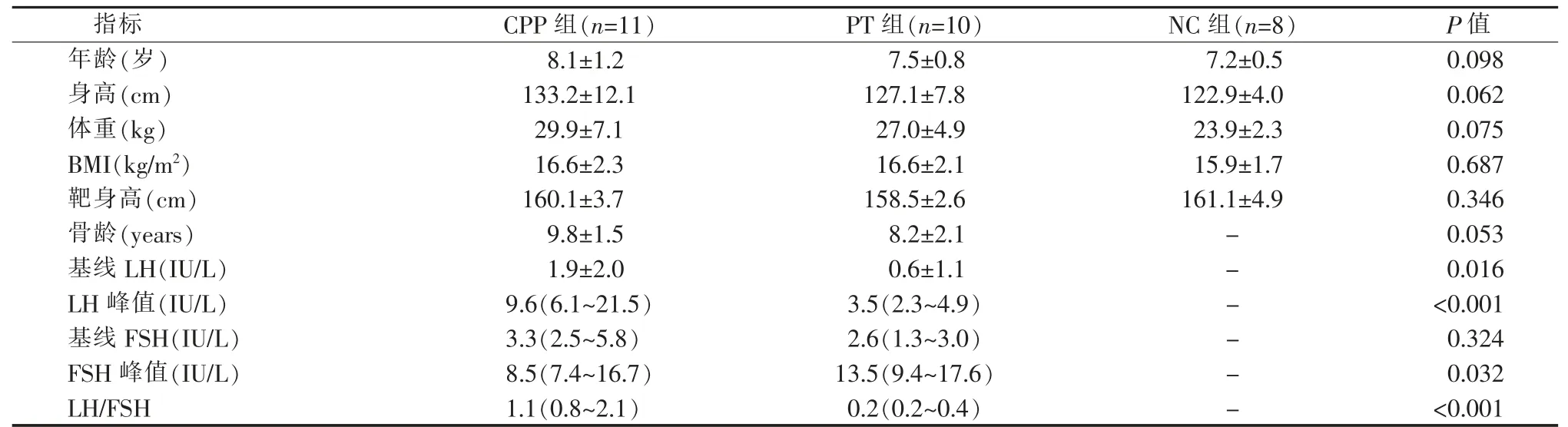
表1 CPP 组、PT 组以及NC 组基线数据特征
二、CPP 组、PT 组以及NC 组相关肠道菌群组成分析
在所有粪便样本中共检测到89 个门、78 个纲、153 个目、345 个科、1 133 个属、5 004 种微生物,其中占主导地位的门主要包括厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)及放线菌门(Actinobacteria)等。CPP 组、PT 组以及NC 组间具有显著差异的门主要为厚壁菌门、拟杆菌门及变形菌门(Proteobacteria)。
1.门水平:基于不同物种丰度表对各组进行了PCA 和NMDS 分析,其物种组成越相似,则它们在PCA 和NMDS 图中的距离则越接近。在门水平行两两组间比较,CPP 组与NC 组间差异有统计学意义(P=0.019),PT 组与NC 组间的差异更显著 (P=0.003),但CPP 组与PT 组间差异无统计学意义(P=0.143);在属水平,依然是NC 组与CPP 组、PT 组间分别存在差异(P 值分别为0.007 和0.001),而CPP组与PT 组间的差异无统计学意义(P=0.314)(见图1)。总之,性早熟女童肠道菌群在门分类水平的组成及丰度上,与健康女童均存在差异。

图1 3 组菌群基于门水平的PCA(A)和NMDS 结果(B)及属水平(C)的PCA 和NMDS(D)结果
在门分类学水平上,尽管3 组的优势菌门均为厚壁菌门、放线菌门和拟杆菌门,但不同组别丰度存在明显差异。NC 组,厚壁菌门和放线菌门的丰度明显高于CPP 组及PT 组 (CPP 组、PT 组及NC 组的厚壁菌门丰度分别50.03%、37.73%及67.61%,而放线菌门丰度分别为4.26%、4.22%及11.16%),而拟杆菌门的丰度低于CPP 组和PT 组 (分别为33.00%、43.26%及10.32%)。由此可见,较健康女童,性早熟女童拟杆菌门丰度升高,厚壁菌门及放线菌门丰度降低(见图2)。
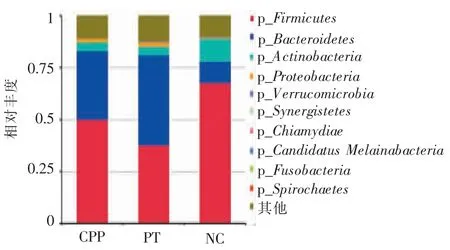
图2 3 组菌群在门(P)分类学水平上的相对丰度
2.属水平:在属分类学水平上,3 组的优势菌属均为拟杆菌属(Bacteroides),但性早熟女童较健康女童的拟杆菌属丰度升高,其中PT 组最高(29.93%),CPP 组次之(24.29%),NC 组最低(7.33%),差异统计学意义(P=0.005)。NC 组放线菌属的丰度明显低于性早熟(CPP+PT)组。此外,粪杆菌属(Faecalibacterium)丰度在CPP 组最高(8.20%),NC 组次之(7.04%),PT 组最低(6.39%);双歧杆菌属(Bifidobacterium) 在3 组间的丰度差异则无统计学意义(P=0.452)(见图3)。
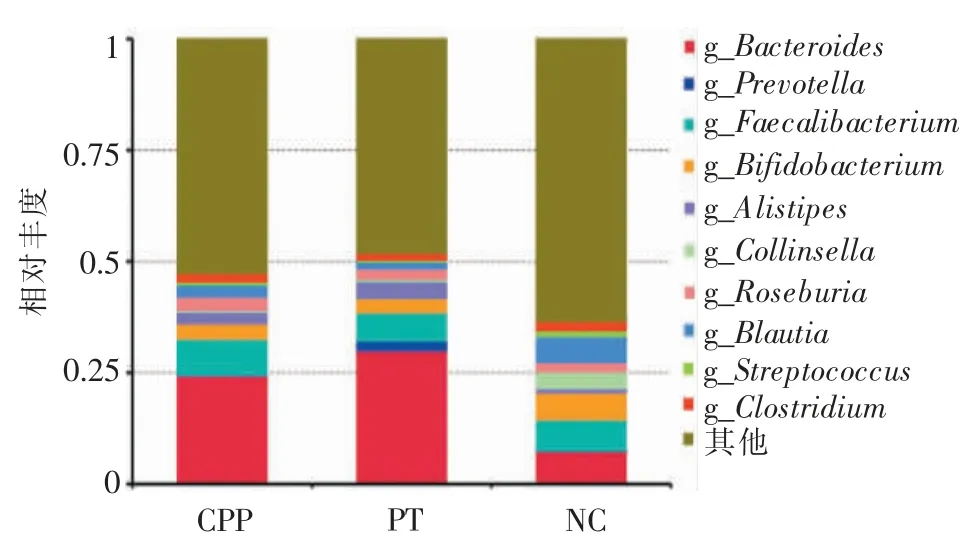
图3 3 组菌群在属(G)分类学水平上的相对丰度比较
3.种水平:在种分类学水平上,3 组的优势菌种均为普氏粪杆菌种(Faecalibacteriumprausnitzii),其丰度分别为5.41%、4.33%及4.75%,同粪杆菌属一样,CPP 组丰度高于PT 组及NC 组。进一步进行Metastat 分析,对3 组肠道菌群的物种丰度进行假设检验得到P 值,对P 值进行校正得到Q 值,根据Q 值筛选3 组间的差异性物种。在种水平上,组间差异显著的前6 位分别为脆弱拟杆菌种(Bacteroides fragilis)、卵形拟杆菌(Bacteroides ovatu)、沙氏拟杆菌种(Bacteroides sartori),单形拟杆菌(Bacteroides uniform)、普通拟杆菌种(Bacteroides vulgatus)及木糖降解拟杆菌(Bacteroides xylanisolvens),其均属于拟杆菌属。其中,脆弱拟杆菌种、卵形拟杆菌、沙氏拟杆菌种在PT 组与NC 组间的差异显著(Q<0.05),普通拟杆菌种在CPP 组与NC 组间的物种差异显著(Q<0.05),单形拟杆菌种及木糖降解拟杆菌种在CPP 组与NC 组、PT 组与NC 组间的差异均显著(Q<0.05)(见图4)。
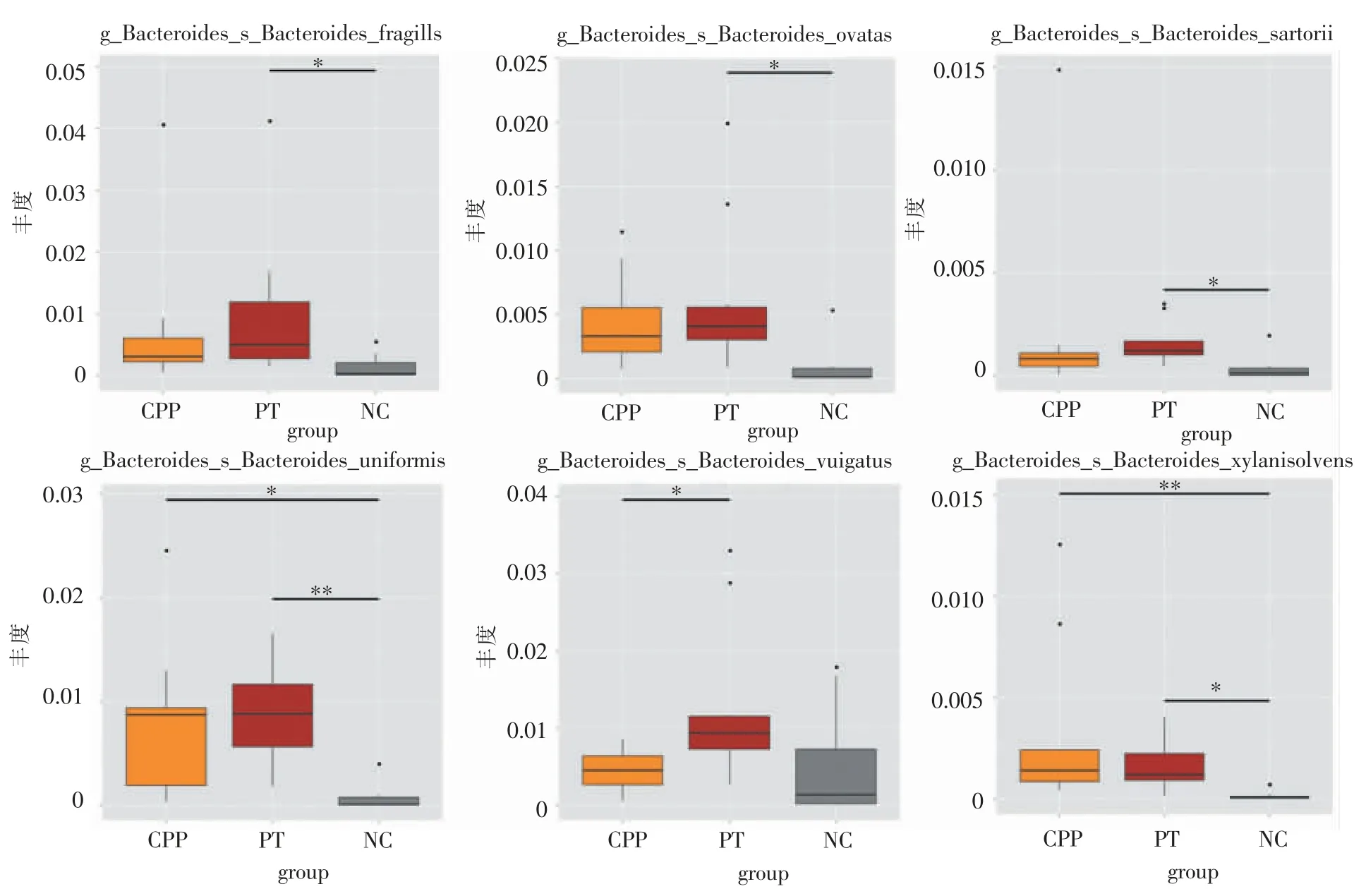
图4 3 组间在种水平有显著差异的物种
4.丰度:CPP 组、PT 组、NC 组间的物种丰度聚类分析显示,在门水平上,CPP 组的互养菌门(Synergistetes)及绿脓杆菌门(Chlorobideferribacteres)显著富集,PT 组的Candidatusomnitrophica、硝化螺旋菌门(Nitrospirae)及产水菌门(Aquificae)富集,NC组的放线菌门及Candidatusmelainabacteria 富集。在属水平上,CPP 组,普拉梭菌属(Faecalibacterium)、罗斯菌属(Roseburia)、霍尔德曼菌属(Holdemanella)及巨球型菌属(Megasphaera)富集;PT 组,颤螺旋菌属(Oscillibacter)及普雷沃菌属(Prevotella)显著富集;NC 组,粪球菌属(Coprococcus)及丁酸球菌属(Butyricicoccus)富集(见图5)。
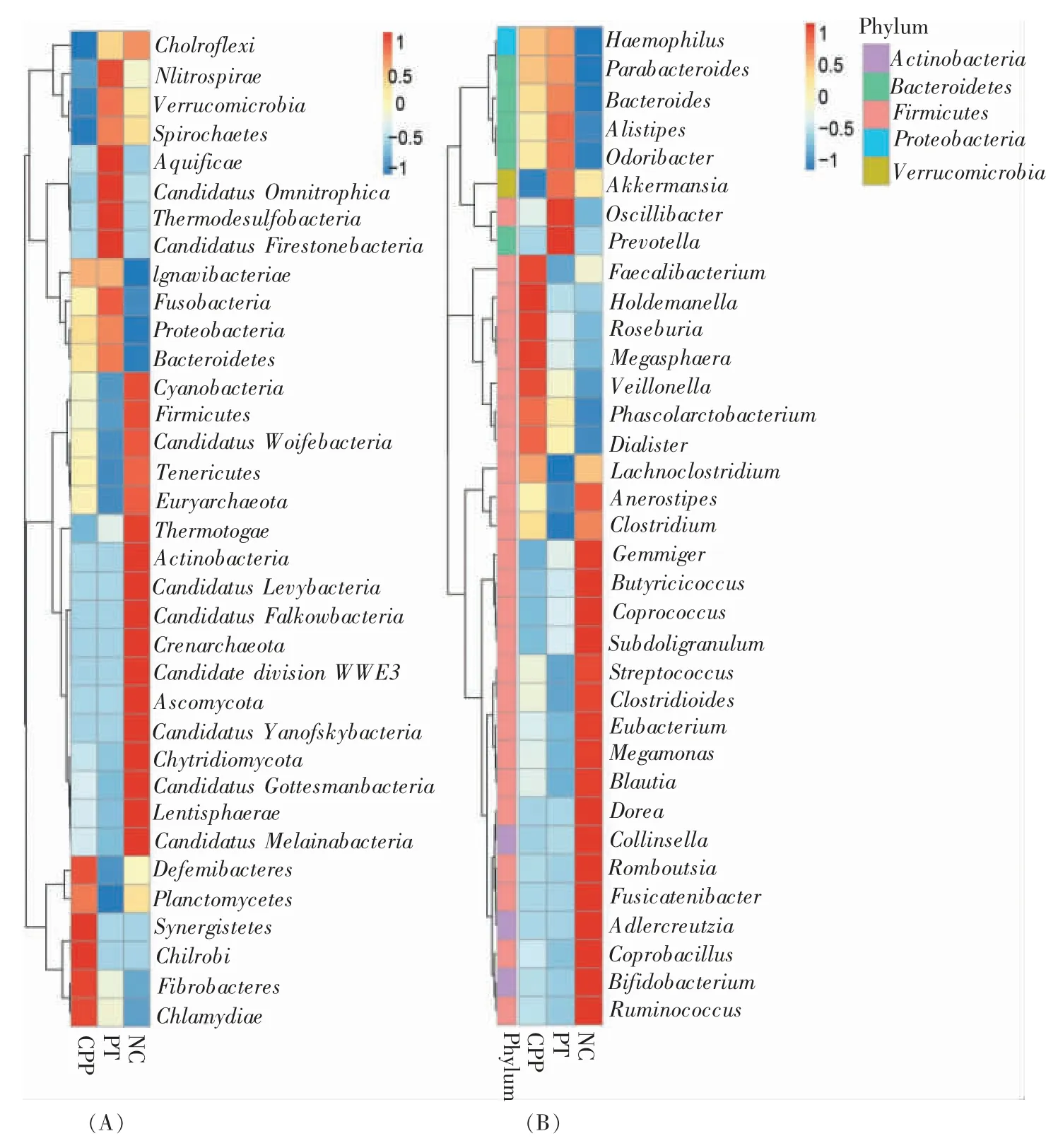
图5 3 组间菌群的物种丰度聚类分析热图(前35 位)
三、CPP 组、PT 组以及NC 组耐药基因分析
本研究在全部29 个粪便样本中均发现了耐药基因。与CARD 库比对,共发现312 个抗性基因,其中性早熟女童所携带的共同耐药基因为298 个,明显多于健康对照组240 个。PT 组抗性基因及ARO数目最多,CPP 组次之,NC 组最少。抗性基因的物种归属图可以看出,CPP 及NC 组中抗性基因含量最多的均为厚壁菌门,其占全部耐药菌的百分比分别为29%及30%,PT 组抗性基因含量较多的主要为拟杆菌门(占全部耐药菌百分比的43%)(见图6)。
基于抗性基因的丰度进行Anosim 分析显示,CPP 组、PT 组与NC 组间差异有统计学意义(R=0.1878,P=0.04),PT 组与NC 组间差异更加显著(R=0.2873,P=0.009)。CPP 组、PT 组及NC 组分别发现有257 种、273 种和240 种耐药基因(见图4)。3 组共发现特异性耐药基因56 种,CPP 组中发现了16 种(主要为β-内酰胺类CAU-1 和ACI-1 以及氨基糖苷类AAC3-Id),PT 组中发现了26 种(主要为四环素类tetS、复合抗生素类ramA 及多肽类basS),NC 组中发现了14 种 (主要为四环素类tetK、大环内酯类Erm34 及OprM)(见图7)。
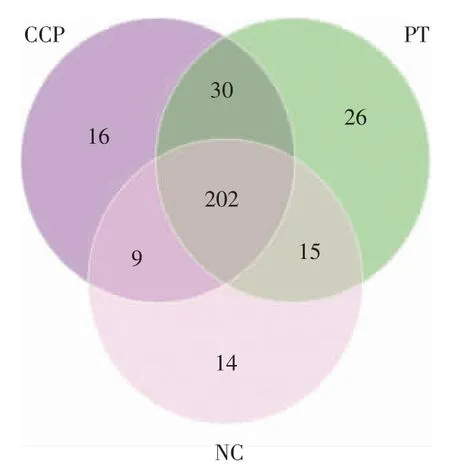
图7 3 组间耐药基因种类数目韦恩图
CPP 组与PT 组抗性基因丰度前3 位均为四环素类抗性基因(tetQ)、大环内酯类抗性基因(ErmF)、喹诺酮类抗性基因(adeF),但PT 组抗性基因丰度明显高于CPP 组。NC 组中糖肽类耐药基因(vanSC)未检出,抗性基因主要集中在四环素类和大环内酯类(见图8)。参考抗性基因的相对丰度表,用单因素方差分析对CPP 组、PT 组及NC 组的肠道菌群抗性基因差异性进行分析。CPP 组、PT 组及NC 组3 组中四环素类抗性基因tetQ 相对丰度分别为141.21、282.03 及37.10,大环内酯类抗性基因ErmF相对丰度分别为128.83、174.88 及51.13。性早熟女童的四环素类及大环内酯类抗性基因丰度远高于健康女童。
利用Metastats 方法筛选具有显著差异的耐药基因。CPP 组与NC 组相比,amrB、catB2、cat86、clbC、OXA_278、MuxB、OCH_7、FosA2、CepS、SMB_1具有显著差异(Q 值均<0.05);PT 组与NC 组相比,tetQ、vanSC、dfrF、CblA_1、vgbB、vanM、vanTC、mtrD、vanWI、Klebsiella_pneumoniae_acrA 组间差异极显著(Q 值均<0.01);CPP 组与PT 组相比,amrB、catB2、cat86、clbC、ramA、MIR_9、MIR_16、vanWI、dfrC、CepS具有显著差异(Q 值均<0.05)。氨基糖苷类抗生素amrB,酚类抗生素catB2、cat86 及clbC 类抗性基因是可能是区别CPP 与PT 及健康女童的标志性抗性基因。
讨 论
一、性早熟儿童肠道菌群与健康儿童存在差异
本研究通过宏基因组测序分析发现,在门水平上,CPP 组及PT 组的优势菌门均为厚壁菌门和拟杆菌门,但CPP 组以厚壁菌门为主,PT 组以拟杆菌门为主,NC 组的优势菌门也为厚壁菌门,但3 组间组优势菌门在丰度上存在显著差异:NC 组厚壁菌门和放线菌门的丰度明显高于性早熟(CPP+PT)组。在属分类学水平上,尽管3 组的优势菌属均为拟杆菌属,但丰度有显著差异,性早熟患儿粪便中拟杆菌属的丰度明显高于正常女童。NC 组的放线菌属丰度最低,而CPP 组的粪杆菌属丰度最高。进一步分析发现,在种水平上有6 种拟杆菌存在明显的组间差异,分别是脆弱拟杆菌种、卵形拟杆菌种、沙氏拟杆菌种、单形拟杆菌种、普通拟杆菌种和木糖降解拟杆菌种。
既往研究发现,肥胖儿童与体重正常儿童相比,肠道中厚壁菌门丰度增加,拟杆菌门丰度降低,厚壁菌门/拟杆菌门比例升高[12],同时多形拟杆菌有抑制肥胖的作用[13]。Dong 等[14]对CPP 女性患儿和健康女童的肠道菌群进行16S rDNA 测序发现,CPP组的肠道菌群与肥胖相关的肠道菌属相似[14]。有趣的是,本研究结果显示,性早熟女童较健康女童拟杆菌门丰度升高,厚壁菌门丰度降低,与Dong 等[14]的研究相反。这可能是由于本研究本样本量较少,也可能由于本研究入组的对象BMI 不存在差异,今后需要扩大样本量并纳入不同BMI 水平的研究对象,需进一步验证该结论。周莎莎等[15]采用16S rDNA 测序研究发现,CPP 组肠道内的粪杆菌属丰度显著增高,与本研究的宏基因组分析结果一致。本研究还发现,CPP 组与NC 组间的粪杆菌属中普氏粪杆菌种存在明显差异。粪杆菌属的增多造成了L-色氨酸的降低,CPP 的发生与粪杆菌属的紊乱影响色氨酸代谢通路有关[15]。性发育不同阶段,肠道菌群具有的生物多样性[16],健康儿童和成年人拥有相似数量的分类群和功能基因,但其组成和功能潜能却存在显著差异。既往研究表明,儿童富集双歧杆菌和粪杆菌,而成年人含有较丰富的拟杆菌[17]。本研究中性早熟女童较健康女童肠道内有更高丰度的拟杆菌,这可能与性发育本身有关,需要进一步对菌群功能进行深入研究。美国学者通过动物实验证实,哺乳期肠道菌群重建可以预防或治疗胰岛素抵抗相关性早熟[18]。而我国研究者比对了CPP、超重与健康女童间的肠道菌群,发现CPP 组肠道菌群的Alistipes、克雷伯菌等显著上升,菌群间相关性增强,表明菌群合成的神经递质可能在CPP 发病中有重要作用,提示靶向肠道菌群或能用于CPP治疗[19]。由此可见,性早熟发病可能与肠道菌群结构显著变化有关,肠道菌群在性早熟的发生、发展过程中可能发挥着重要作用。
二、宏基因组测序技术检测提示性早熟女童肠道菌群抗性基因富集
耐药基因不仅广泛存在于环境中,也存在于人体肠道系统中。宏基因组测序技术具有特异性高,灵敏度高,高通量的特点[20],相较于传统的微生物的培养和筛选过程,该方法可以检测环境微生物的抗生素抗性组(antibiotic resistome),即微生物中所有抗性基因的集合,包括不表达或低表达的抗性基因,以及具有较低抗性或与抗生素密切相关,有可能进化为抗性基因的耐药基因前体[21]。有研究对11 国180 名成年受试者的粪便进行宏基因组检测,发现健康成人肠道菌群的细菌群落结构与耐药基因谱图显著相关。四环素类、大环内酯类、林可酰胺类耐药基因是目前3 种最丰富的抗性基因类型[22-23]。中国人群肠道中抗性基因丰度最高,大环内酯类ermF 可作为中国人肠道菌群中抗性基因的标志。与成人不同的是,在本研究中健康女童肠道菌群抗性基因最丰富前3 位分别为四环素类、大环内酯类及氨基糖苷类抗性基因,而性早熟女童与健康女童的抗性基因组成有着显著差异,且更加富集于四环素类及大环内酯类抗性基因。芬兰的学者观察了儿童的肠道菌群及抗生素使用情况,发现大环内酯类抗生素的使用与其耐药性之间,在遗传和表型水平上可能存在因果关系,会增加儿童肥胖风险。大环内酯类抗生素的使用使双歧杆菌(Bifidobacteria)及克里斯滕森菌科(Christensenellaceae)丰度降低,拟杆菌属(Bacteroides)和丹毒丝菌属(Erysipelotrichaceae)丰度升高。这些菌群被认为与儿童或成人的肥胖及代谢性疾病有关[24],其四环素同时能够促进脂肪堆积。
我国的学者研究发现,长期暴露于低剂量抗生素可能是儿童肥胖的一个风险因素,接触甲氧苄啶、氟苯尼考和畜禽类抗生素水平较高的受试者有较高肥胖风险,这激励研究者进一步探索抗性基因在肥胖和性早熟共同发生中的作用[8]。本研究在所有儿童粪便样本中均发现了耐药基因,同时还从儿童体内检测出临床已经停用多年和儿童慎用及禁用但在环境和食品中经常发现的抗生素抗性基因,如喹诺酮类、四环素类。已有研究发现,新生儿粪便中已经含有β-内酰胺类和四环素类抗性基因[25],从儿童、青少年到成人肠道内抗性基因的多样性随年龄的增长而增加[26],可见抗生素滥用不仅是临床治疗的问题,环境和食品也是儿童抗生素的重要暴露源,目前全球超过一半以上的抗生素用于畜禽养殖[27],畜禽使用抗生素可能主要通过污染水及食物进入儿童体内。
抗生素的使用是一把双刃剑。研究表明,多种抗生素在亚治疗剂量水平具有促生长作用,曾经广泛应用于农业和畜禽业[28-29]。在青春发育前,抗生素对儿童同样有促生长作用[30]。抗生素作为生长促进剂的作用机制是通过其抗菌特性破坏肠道菌群导致[26]。有动物实验证明,无菌动物体内由于缺乏抗菌生长促进剂,从而导致其生长减缓[31]。细菌由于携带抗性基因而对抗生素产生耐药性,使其对抗生素产生了抵抗能力,同时造成肠道菌群的紊乱,有可能进一步通过肠道菌群-肠-脑轴影响中枢神经系统的功能,从而影响生长、发育。有证据证明,抗性基因进入环境或病原微生物中通过水平转移在细菌间传播,越来越多的抗性基因在畜禽养殖环境及动物肠道中被发现[32],可能通过食物链传递给人类,给儿童健康带来潜在的风险[33]。
本研究存在一定局限性。首先,因为鸟枪法宏基因组测序技术的成本相对比较昂贵,所以本研究纳入的总样本量相对有限。其次,在纳入样本时,缺少针对出生史、喂养史等因素的分层分析,分娩及喂养方式的不同有可能会导肠道菌群改变。最后,耐药基因的分布具有一定的区域性特征,在低收入、不发达地区人群的肠道菌群中抗性基因的丰度要高于生活在发达地区的人群[22],而本研究纳入的对象均来自于长三角地区,饮食结构及经济基础可能与其他区域有一定区别。
尽管存在这些局限性,但本研究是首次利用宏基因组测序技术,分析性早熟及健康女童的肠道菌群及抗性基因的研究,将为理解肠道菌群及抗性基因对儿童生长、发育的影响及作用迈出重要的一步。由于肠道菌群的复杂性和多样性,其诱导性早熟的机制仍有待进一步研究。未来的研究将基于更大样本量的数据分析,以阐明肠道菌群-肠-脑轴与性早熟之间的关联机制,确定与性早熟相关的肠道菌群的潜在致病成员,以及性早熟患儿个体的特定肠道菌群管理,并建立基于肠道菌群对性早熟的干预和治疗策略。
本研究使用宏基因组测序技术,分析比较了性早熟与健康女童间的肠道菌群结构及组成,发现其存在显著差异,提示性早熟发病可能与肠道菌群结构显著变化有关。同时,本研究发现性早熟女童与健康女童间的抗性基因组成有着显著差异,且更加富集于四环素类及大环内酯类,还从儿童体内检测出临床已经停用多年和儿童慎用及禁用的抗性基因,氨基糖苷类抗生素的amrB,酚类抗生素的catB2、cat86、clbC 类抗性基因是可能是CPP 女童区别与PT 及NC 女童的标志性抗性基因。有关抗性基因对儿童肠道菌群的影响,目前依然知之甚少,且尚没有证据可以证明动植物养殖及环境中抗性基因与性早熟的发生、发展直接相关,但研究者不能忽视抗性基因通过可移动的质粒传递给儿童的潜在威胁。
本研究呼吁研究者更多地关注儿童饮食与环境健康,以及环境中抗生素的使用对性早熟发病的潜在威胁,需进一步探索抗性基因在环境中的传播机制以及对儿童健康的影响。
利益冲突声明:所有作者均声明不存在利益冲突关系。
猜你喜欢
当代水产(2022年7期)2022-09-20
西南农业学报(2022年3期)2022-04-25
家庭百事通·健康一点通(2020年8期)2020-09-08
学生导报·东方少年(2020年1期)2020-05-06
植物保护(2019年2期)2019-07-23
赢未来(2018年23期)2018-12-20
家庭百事通·健康一点通(2018年4期)2018-05-16
现代农业科技(2009年19期)2009-03-20
