
紫色球杆菌视紫红质光谱特性的机器学习研究
2022-06-02 08:14郏丽丽孙婷婷
浙江大学学报(理学版) 2022年3期
郏丽丽,孙婷婷
(浙江科技学院 理学院,浙江 杭州 310023)
紫色球杆菌视紫红质光谱特性的机器学习研究
郏丽丽,孙婷婷*
(浙江科技学院 理学院,浙江 杭州 310023)
近年来,机器学习等人工智能技术被应用于蛋白质工程,其在蛋白质结构、功能预测、催化活性等研究中具有独特优势。在未知蛋白质结构的情况下,将蛋白质序列和功能特性与机器学习相结合,基于序列-活性关系(innovative sequence-activity relationship,ISAR)算法,将蛋白质氨基酸序列数字化,用快速傅里叶变换(fast four transform,FFT)进行预处理,再进行偏最小二乘回归建模,可在数据集较少情况下拟合得到最佳模型。通过机器学习对紫色球杆菌视紫红质(gloeobacter violaceus rhodopsin,GR)的突变体蛋白质氨基酸序列与光谱最大吸收波长进行建模,获得了最佳模型。用最佳索引LEVM760106建模得到的确定系数R2为0.944,均方误差E为11.64。用小波变换进行的预处理,其R2虽也约为0.944,但E大于11.64,不及FFT进行的预处理。方法较好地解决了蛋白质序列与功能特性之间的数学建模问题,在蛋白质工程中可为预测更优的突变体提供支持。
机器学习;数字信号处理;光谱特性

机器学习是数理统计和计算机科学的延伸,包含许多统计模型和计算机程序算法。1992年,MUGGLETON等[1]运用机器学习算法预测蛋白质二级结构,将蛋白质的二级结构与机器学习的编码相关联。随着机器学习算法的逐渐成熟,出现了用于预测结构、折叠、结合甚至催化活性的新版本机器学习算法,其主要目的是处理有关突变体及其功能特性的积累信息。易华伟等[2]基于蛋白质的氨基酸序列通过机器学习算法预测其稳定性。程淑萍等[3]运用3种不同的机器学习算法预测非编码RNA和蛋白质之间的相互作用,提高预测的准确率。徐开琨等[4]运用特征选择与机器学习算法研究生物蛋白质中的标志物,这对疾病的早期诊断和临床治疗等有重要意义。胡如云等[5]介绍了机器学习在合成生物学领域的典型应用,如启动子预测、酶催化设计、代谢途径构建、基因线路设计等。
然而机器学习算法大多要求大数据集,只有在大量数据下结合其生物化学特性,才能拟合得到较优的模型,更好地开展定向进化的蛋白质研究工作[6-7]。曲戈等[8-9]通过蛋白质定向进化,产生大量组合突变体数据集,运用机器学习算法较好地获得了定制化蛋白质。MOSELEY[10]通过将蛋白质的生物化学特性表达与机器学习算法相结合,构建出鲁棒性好的统计模型,预测未知的改良突变体。因此,机器学习算法在蛋白质改造研究中有重要应用。
以上机器学习算法都是基于大数据集进行的蛋白质工程研究,在只有少量数据集下进行的机器学习算法研究较少。同时,大多研究基于蛋白质的结构,对未知结构的蛋白质分子及其功能特性的研究也很少。基于序列-活性关系(innovative sequence-activity relationship,ISAR)[11]的算法,为蛋白质工程提供了新的研究方向。
ISAR算法解决了数据集少和未知蛋白质结构的问题,有助于探究蛋白质序列和功能特性的相互关系。ISAR算法由FONTAINE等[12]于2018年开发,以氨基酸的物理化学性质为基础,用光谱表示蛋白质。CADET等[13]解释了ISAR算法在改善来自黑曲霉的环氧化物水解酶对映选择性中的应用。该实验基于9个单点突变(29)的组合,预测了这512个变异体的对映选择性,发现了具有更高对映选择性的突变体。并运用ISAR算法对4个不同类型的数据集(热稳定性的细胞色素P450、结合亲和力的TNF-alpha、结合亲和力的GLP-2和热稳定性的肠毒素)进行拟合建模,验证了ISAR算法能有效捕捉突变对蛋白质功能的影响。NICOLAS等[14]提出了用不同组合编码扩展序列的方法建模,测试了4种不同长度的蛋白质(GLP-2、TNFalpha、细胞色素P450和环氧水解酶)及其活性(cAMP激活、结合亲和力、热稳定性和对映选择性),并预测了一个多肽链的适应度值。OSTAFE等[15]通过不同pH的不同介质用ISAR算法提高了葡萄糖氧化酶的活性,使葡萄糖氧化酶突变体(P1)对介质二茂铁-甲醇(12倍)和硝基苯胺(8倍)具有更大的特异性,并在3个pH调整缓冲区中表现出更好的性能,在存在二茂铁甲醇的情况下,与pH为5.5的野生型酶相比,P1的kcat/KM比率增加了121倍。
质子泵型视紫红质(proton-pumping rhodopsin,PPR)[16]蛋白是一种结构比较简单且广泛存在的光能转换系统。结合视黄醛分子在吸收光子后令其构象改变,引起整体蛋白的变构等,在一定程度上能促进三磷酸腺苷酶(adenosine triphosphate,ATP)的合成,将光能转化为化学能[17-18]。PPR通过将光能转化为化学能,从而提高细胞的积累生物量。紫色球杆菌视紫红质(gloeobacter violaceus rhodopsin,GR)是一种PPR,通过取代GR的不同氨基酸获得不同的GR突变体,影响其吸收光谱的峰值。吸收光谱的峰值越大,光能转化率越高,微生物的生产和细胞的耐受性越强。因此,需对GR光谱特性进行研究。
通过对GR视网膜结合口袋进行视紫红质的氨基酸替换,组合成新的GR突变体,以调节其光谱最大吸收波长λmax[19]。从数据集中提取81条少量GR突变体,用ISAR算法对数据进行数字信号处理(digital signal processing,DSP),再与实验获得的最大光谱吸收波长进行偏最小二乘回归(partial least squares regression,PLSR)建模,采用交叉验证方法获得符合GR实验数据要求的最佳模型和蛋白质氨基酸序列与光谱特性的关系。
1 ISAR算法
ISAR是一种创新的序列-活性关系算法,基于快速傅里叶变换(fast Fourier transform,FFT)[20]等DSP算法,将实验与蛋白质计算设计相结合。ISAR算法分编码、建模和预测3个阶段。图1为ISAR算法流程。
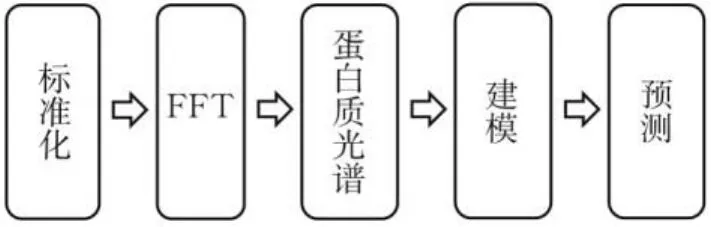
图1 ISAR算法流程Fig.1 The flow of ISAR methodology
1.1 编码阶段
在氨基酸指数索引(AAindex)数据库中找到符合蛋白质特性的最佳索引。AAindex是含566组索引数字指数的数据库,每组索引包括20种标准氨基酸,代表各自的物理化学和生物化学性质,同时给出了这些索引之间的相关性及其他信息[21]。依据AAindex数据库,将蛋白质的氨基酸序列数字化,再用FFT将数字信息转换为能量对频率的蛋白质光谱形式。其可通过
实现,其中,N为输入信号的长度,S为输出光谱(复数),k为光谱的频率,s(n)为有限长连续信号,n为输入信号的位置,i为虚数单位。
DSP是一种数据预处理技术,对信号进行分解和处理,以不同方式进行数据表达。FFT是DSP的一种方法,BENSON[22]很早就将傅里叶变换方法应用于生物序列的比较。FFT中一个点的变换将影响整条数据的光谱,因此蛋白质位点中任何一个突变点的变化,都会令蛋白质光谱整体变换,这也是ISAR算法的亮点之一。已有很多方法研究单点突变的变换,将每个特性值累加,忽略了氨基酸之间的相互作用。
1.2 建模阶段
对蛋白质光谱与蛋白质突变体的生物化学特性值进行PLSR建模,并用留一交叉验证法(leave-one-out cross validation,LOOCV)验证模型和提高模型的鲁棒性。蛋白质的生物化学特性值除通过实验获得外,还可将生物、数学和计算机相结合,通过机器学习拟合蛋白质的数据预测得到。对无法获得晶体结构的蛋白质,通过已知蛋白质的氨基酸序列特征属性,用ISAR算法便可获得蛋白质的生物化学特性值。
由机器学习算法进行建模,通常需要大量数据集进行训练和验证。在ISAR算法中,可用PLSR解决数据集小的问题。为防止模型过拟合,通过LOOCV优化模型参数和验证模型,同时用确定系数R2和均方误差E(式(2)和(3))衡量模型好坏。LOOCV是交叉验证法的一种,由于样本数n较少,将n-1个样本作为训练集,留出1个样本作为测试集,循环n次,共可获得n个测试数据。
根据AAindex数据库中566组索引与实验数据进行建模,获得多个模型。在同一组索引下,由不同参数得到若干个不同模型。在参数不变的情况下,566组索引通过LOOCV得到n×566个模型。不同参数和不同组索引得到不同的确定系数R2和均方误差E。根据最小E和较优R2找到最佳索引,从而获得最佳模型。
1.3 预测阶段
将建模阶段选出的最佳索引经实验数据预处理后放入模型,预测与实验数据相对应的特征属性值,同时验证氨基酸序列与蛋白质功能属性的相关性。因此,使用有限的实验数据通过ISAR算法拟合可获得适应其特征属性的最佳模型,进一步预测其他可能存在的更好的突变体。
2 结果与讨论
2.1 实验数据
GR通过定向进化获得81条突变体吸收波长数据集,包含单点突变和组合突变(位点:W122,V126,M158,G162,E166,G178,S181,F185和A256)[23]。GR的野生型光谱最大吸收波长λmax为539 nm,通过替换不同的氨基酸,获得的突变体的λmax为50~650 nm。因此,组合突变体和氨基酸之间的相互作用会影响GR的λmax。
数据预处理是建模前的关键步骤。首先将81条数据进行氨基酸编码,根据AAindex数据库,每组索引有20种不同的氨基酸,每种氨基酸用不同的数值代替。GR野生型(wide type,WT)是由20个标准氨基酸组成的长为298的氨基酸序列。首先根据AAindex数据库进行数值化处理,然后由FFT转换为蛋白质光谱。图2为GR的WT、单点突变体W122A和组合突变体G162L/E166W/F185A/A256S的编码过程。其他突变体的光谱转换类似。W122A为GR的第122个位点,是色氨酸(tryptophan,W)突变成丙氨酸(alanine,A)的单点突变体。组合突变体G162L/E166W/F185A/A256S由4个位点突变成其他氨基酸得到。3种蛋白质通过FFT得到蛋白质光谱,图3分别为野生型、突变体W122A和组合突变体G162L/E166W/F185A/A256S在不同频率下的振幅。由图3可知,虽然W122A为单点突变、G162L/E166W/F185A/A256S只有4个位点突变,但影响的是整个蛋白的光谱振幅,与WT得到的蛋白质光谱完全不同,这也是ISAR算法的优势。因此,81条数据通过FFT得到完全不同的81条蛋白质光谱,为后续的吸收光谱建模打下良好基础。
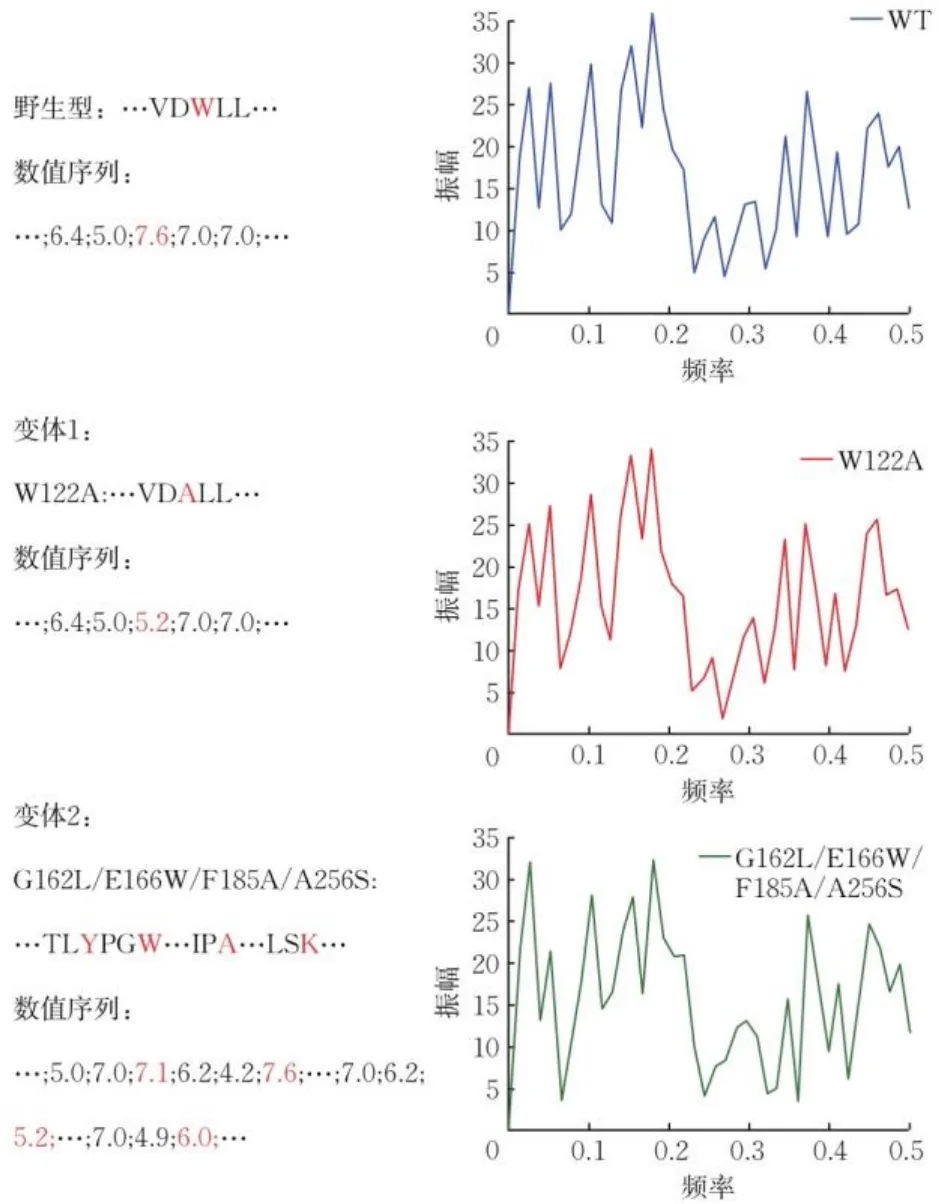
图2 用ISAR算法由GR数据得到的不同蛋白质光谱Fig. 2 Different protein spectra are obtained from GR data by ISAR method
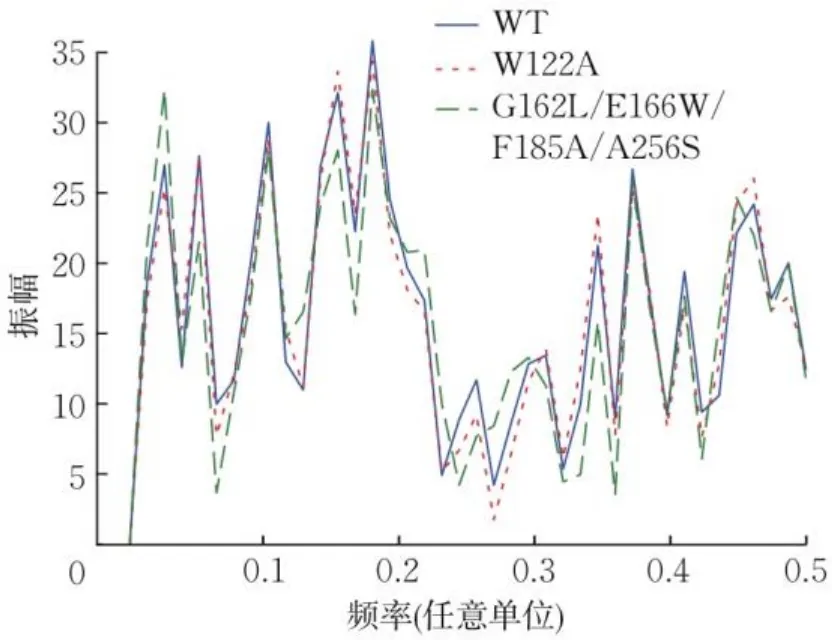
图3 3个蛋白质通过FFT转变为蛋白质光谱Fig.3 Three proteins are transformed into protein spectra by FFT
2.2 选择最佳索引
GR是由20种标准氨基酸组成的蛋白质,氨基酸的数值化和相互作用均会影响GR的吸收光谱特性。AAindex数据库有566组氨基酸索引,其中16组因部分氨基酸数值缺失被剔除。利用剩下的550组索引在固定参数下与PLSR建立模型,并测试其E值,如图4所示。不同索引的E值不同,550组索引的E值差异较大,对模型的建立和预测有很大影响。由式(2)和式(3)计算R2和E,用较优R2和最小E选择一组最佳索引[24]。首先,编码81条GR突变体氨基酸序列数据,并根据FFT得到81条不同蛋白质光谱。然后,与GR的吸收光谱λmax进行建模,优化模型参数,并用LOOCV得到最佳E和R2。表1为由不同的索引与GR数据建模得到的E和R2以及索引的相关信息。对GR实验数据,用全部的550组索引和不同的模型参数进行模拟,得到的最小E为11.64,与之对应的R2为0.944,最佳索引为LEVM760106。从表1中也可看出,不同索引的E和R2都不同,模型的优劣程度也不同。CEDJ970104索引的R2为0.800,E为20.75,虽然R2相对较高,但E太大,模型拟合度较差,因此舍弃。而CHOC760104和FINA910102索引的R2太小,E更大,模型拟合差,直接舍弃。最终选择的最佳索引为LEVM760106,将其进行GR突变体氨基酸数字化并预测λmax。
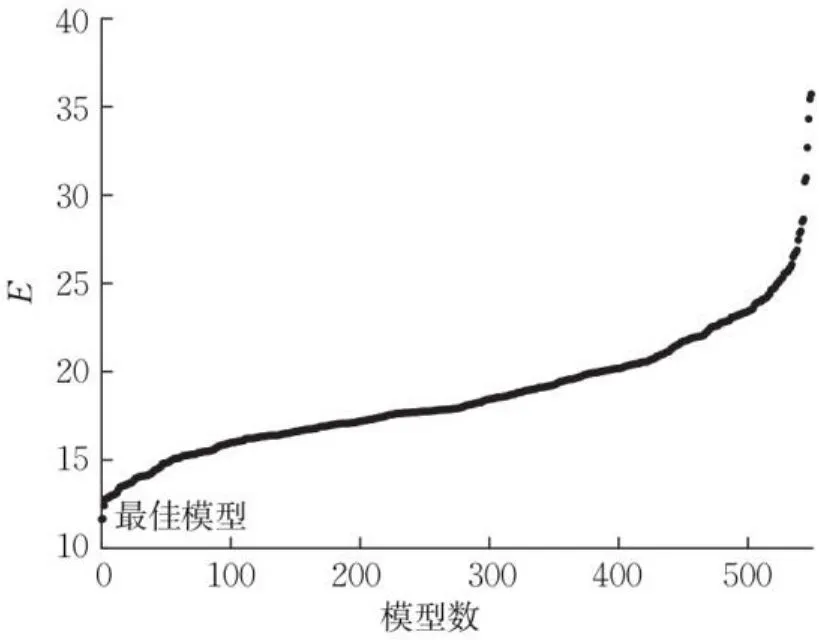
图4 根据不同参数获得550个λmax模型的E值Fig.4 The E ofλmax 550 models according to different parameters
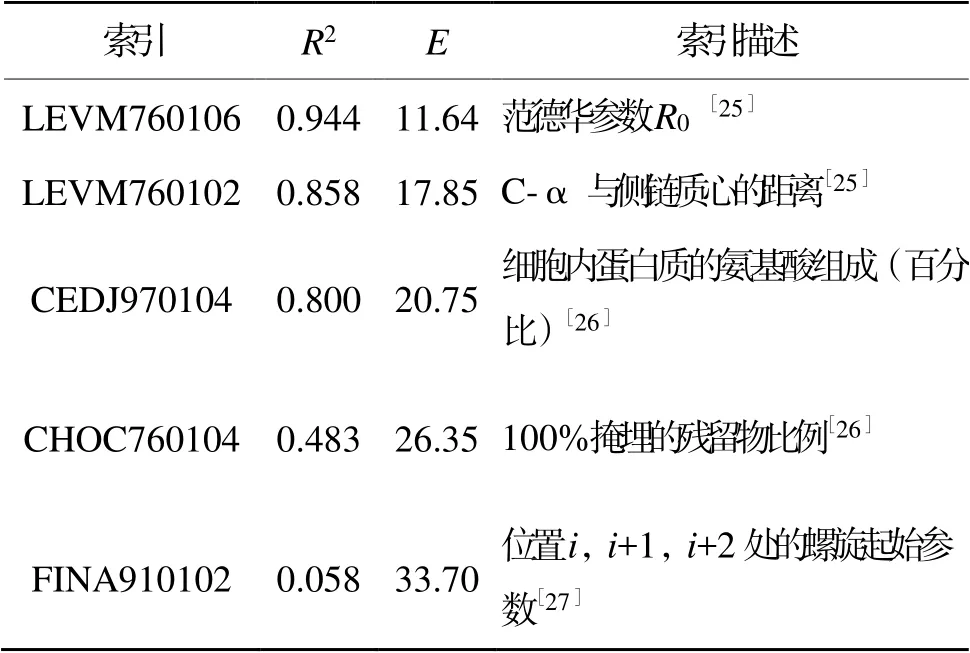
表1 不同索引下的R2和ETable 1 The R2and E under different indexes
2.3 建模和预测
由最小E选择最佳索引LEVM760106,选出最佳模型参数,进行建模与预测。运用ISAR算法预测GR的81条实验数据,得到λmax的实验值和预测值、E=11.64、确定系数R2=0.944,模型的拟合度很高。E和R2是衡量模型优劣的标准,E用于选择最佳索引和最佳模型,R2体现模型的预测能力。由图5知,λmax的预测值和实验值在直线附近聚集,偏离不大,模型鲁棒性较好。
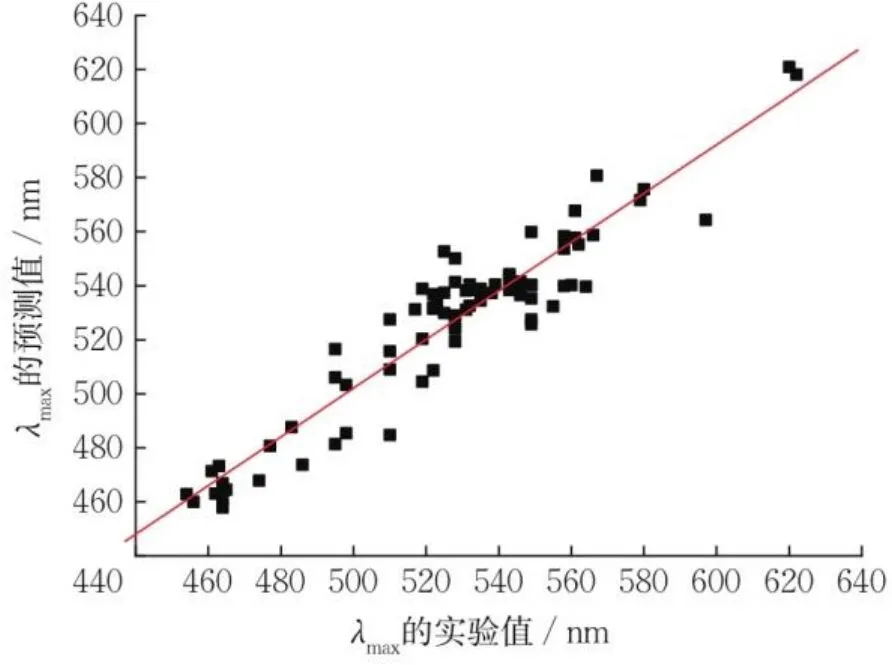
图5 R2= 0.944时GR及突变体的LOOCV预测Fig. 5 Prediction of GR and mutants by LOOCV when R2=0.944
2.4 方法比较
ISAR算法中的一个重要环节是用FFT进行数据处理。FFT将单个位点或多个位点的突变体转换为影响整个蛋白质的变换,改进了以往生物特性功能单纯累加的缺陷。同时本研究也运用小波变换预处理数据,得到的结果如表2所示。小波变换也是DSP的一种方法,是对短傅里叶变换的继承和发展,通过时间或空间的局部变换,突出某地方的特征。
除用LOOCV外,还用十折交叉验证法验证模型的优劣。十折交叉验证法是将样本数n分成10份,选择9份作为训练集,1份作为验证集。通过不同的数据预处理和不同的交叉验证方法,得到不同的E和R2。E越小、R2越接近于1,模型的拟合度越高、鲁棒性越好。由表2可知,用FFT选择的最佳索引均为LEVM760106,且十折交叉验证法增加了验证集数量,令E和R2均有降低。小波变换采用的小波基为db1,得到的E值均较FFT方法大。虽然用小波变换和LOOCV得到的R2与ISAR算法的相差不多,但是E值高了约0.5,模型拟合度不及ISAR算法。实验证明ISAR算法中的FFT非常适合GR的λmax拟合,可获得较优模型,且方便后期的拟合和预测。由表2可知,用4种方法寻找最佳索引,3种方法得到的最佳索引均为LEVM760106。
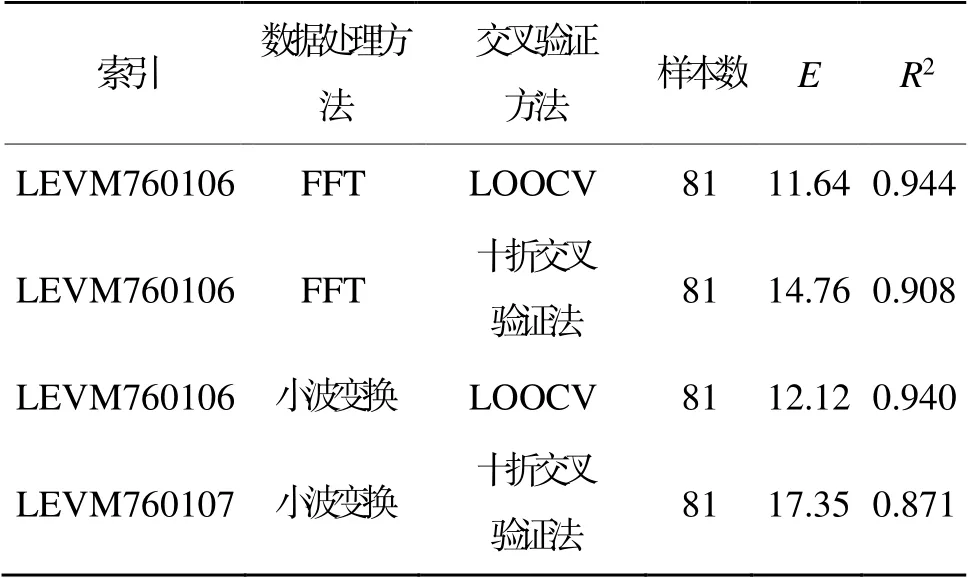
表2 不同方法对GR的验证结果Table 2 Verification results of GR by different methods
表3为最佳索引LEVM760106的20种标准氨基酸的数值表示,每个数值均根据某种物理化学特性、氨基酸之间的相互作用以及公式获得。LEVM760106是关于范德华参数R0[25](一种快速模拟蛋白质折叠)的蛋白质构象简化表示法。从最佳索引LEVM760106的物理化学性质看,突变前后紫色球杆菌视紫红质蛋白质氨基酸序列与光谱吸收波长的相关性与其范德华参数R0密不可分。氨基酸序列、最佳索引和物理化学特性不同,蛋白质和模型参数亦不同。
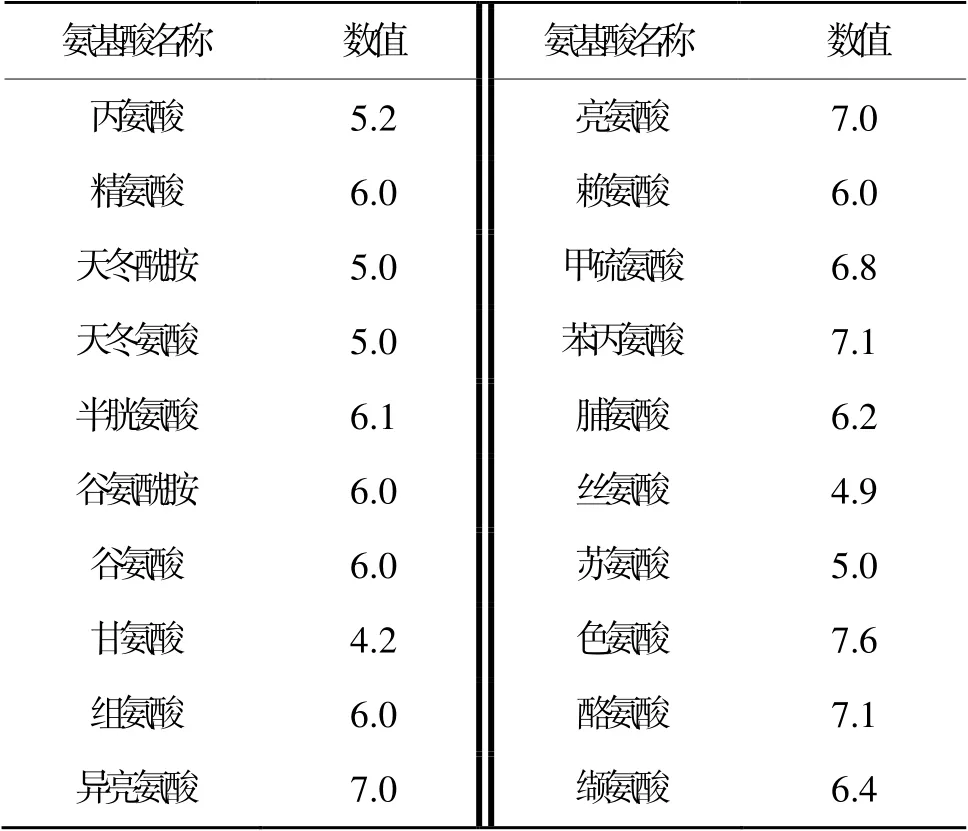
表3 索引LEVM760106中20种氨基酸的数值表示Table 3 Different values of 20 amino acids expressed in index LEVM760106
3 结论
运用AAindex数据库中最佳索引的数字化蛋白质氨基酸序列,用FFT预处理得到影响整体蛋白质光谱的部分变化,与GR的光谱最大吸收波长λmax进行PLSR建模,得到拟合度较高的模型。最佳模型选取的索引为LEVM760106,其中确定系数R2为0.944,均方误差E为11.64。由该索引的物理化学性质,可得到在突变前后的紫色球杆菌视紫红质蛋白序列中,其范德华参数R0变化所引起的物理化学性质与吸收光谱波长的相关性最大。这证明了经傅里叶变换后的频率(蛋白质谱)可有效描述和预测氨基酸序列的蛋白质活性(序列-活性关系用蛋白质谱建模)。此外,此频率考虑了突变对光谱的整体影响,而不是局部影响。ISAR算法的优点之一是不依赖于蛋白质的空间结构,只要获得一定长度的蛋白质氨基酸序列就可与蛋白质的生物化学特性进行建模。优点之二是考虑了氨基酸之间的相互作用力,而不是单纯功能属性值的累加。另外,ISAR算法中的LOOCV和PLSR有效解决了机器学习中数据集少的问题,在数据集量较少时也可进行机器学习。
研究发现,统计预测模型基于氨基酸的物理化学特性建立,并研究了蛋白质氨基酸序列与功能特性的相互关系。用基于已有数据的ISAR算法做预测能否得到比实验数据更好的突变体有待进一步研究。如能通过ISAR算法预测更好的突变体,这将大大节约生物化工实验时间,降低实验成本。统计模型的建立依赖于机器学习,可以预见,机器学习算法将适应更多的生物工程需求,应用于更广泛的生物化工领域。
[1]MUGGLETON S, KING R D,STENBERG M J E. Protein secondary structure prediction using logic-based machine learning[J]. Protein Engineering, 1992, 5(7):647-657. DOI:10.1093/protein/5.7.647
[2]易华伟,唐晓峰. 基于氨基酸序列和模拟结构预测蛋白质稳定性的研究进展[J]. 生物技术通报, 2017,33(4): 83-89. DOI:10.13560/j.cnki.biotech.bull. 1985.2017.04.011
YI H W, TANG X F. Research progress on the prediction of protein stability based on amino acid sequence and simulated structure[J]. Biotechnology Bulletin, 2017, 33(4):83-89. DOI:10.13560/j.cnki.biotech.bull.1985.2017.04.011
[3]程淑萍,谭建军,门婧睿. 基于机器学习方法的非编码RNA-蛋白质相互作用的预测[J]. 北京生物医学工程,2019, 38(4):353-359. DOI:10.3969/j.issn. 1002-3208.2019.04.004
CHENG S P, TAN J J,MEN J R. Prediction of ncRNA-protein interactions based on machine learning methods[J]. Beijing Biomedical Engineering,2019, 38 (4):353-359. DOI:10.3969/j.issn.1002-3208.2019.04.004
[4]徐开琨,韩明飞,黄传玺,等. 基于质谱的蛋白质生物标志物发现中的特征选择与机器学习方法研究进展[J]. 生物工程学报,2019, 35(9):1619-1632. DOI:10. 13345/j.cjb.190064
XU K K, HAN M F,HUANG C X, et al. Research progress of feature selection and machine learning methods for mass spectrometry-based protein biomarker discovery[J]. Chinese Journal of Biotechnology, 2019,35(9): 1619-1632. DOI:10. 13345/j.cjb.190064
[5]胡如云,张嵩亚,蒙海林,等. 面向合成生物学的机器学习方法及应用[J]. 科学通报, 2021,66(3): 284-299. DOI:10.1360/TB-2020-0456
HU R Y, ZHANG S Y,MENG H L, et al. Machine learning for synthetic biology: Methods and applications[J]. Chinese Science Bulletin, 2021,66(3): 284-299. DOI:10.1360/TB-2020-0456
[6]HAMMER S C, KNIGHT A M,ARNOLD F H. Design and evolution of enzymes for non-natural chemistry[J]. Current Opinion in Green and Sustainable Chemistry, 2017,7: 23-30. DOI:10. 1016/j.cogsc.2017.06.002
[7]CHOI Y H, KIM J H,PARK B S, et al. Solubilization and iterative saturation mutagenesis of α1,3-fucosyltransferase from helicobacter pylori to enhance its catalytic efficiency[J]. Biotechnology and Bioengineering, 2016,113(8): 1666-1675. DOI:10. 1002/bit.25944
[8]曲戈,朱彤,蒋迎迎,等. 蛋白质工程:从定向进化到计算设计[J]. 生物工程学报, 2019,35(10):1843-1856. DOI:10.13345/j.cjb.190221
QU G, ZHU T,JIANG Y Y, et al. Protein engineering:From directed evolution to computational design[J]. Chinese Journal of Biotechnology, 2019,35(10):1843-1856. DOI:10.13345/j.cjb.190221
[9]蒋迎迎,曲戈,孙周通. 机器学习助力酶定向 进化[J]. 生物学杂志,2020, 37(4):1-11. DOI:10.3969/j.issn.2095-1736.2020.04.001
JIANG Y Y, QU G,SUN Z T. Machine learning assisted enzyme directed evolution[J]. Journal of Biology, 2020, 37(4):1-11. DOI:10.3969/j.issn. 2095-1736.2020.04.001
[10]MOSELEY L G. Introduction to machine learning[J]. Engineering Applications of Artificial Intelligence, 1988,1(4): 334. DOI:10.1016/0952-1976(88)90057-7
[11]CADET F, FONTAINE N,LI G Y, et al. A machine learning approach for reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes[J]. Scientific Reports, 2018, 8(1):16757. DOI:10. 1038/s41598-018-35033-y
[12]FONTAINE N, CADET F. Method and electronic system for predicting at least one fitness value of a protein,related computer program product: U.S. Patent Application 15/565,893[P]. 2018-04-05.
[13]CADET F, FONTAINE N,VETRIVEL I, et al. Application of fourier transform and proteochemometrics principles to protein engineering[J]. BMC Bioinformatics, 2018,19(1): 382. DOI:10.1186/s12859-018-2407-8
[14]FONTAINE N, CADET F,VETRIVEL I. Novel descriptors and digital signal processing-Based method for protein sequence activity relationship study[J]. International Journal of Molecular Sciences, 2019,20(22):5640. DOI:10.3390/ijms20225640
[15]OSTAFE R, FONTAINE N,FRANK D, et al. One-shot optimization of multiple enzyme parameters: Tailoring glucose oxidase for pH and electron mediators[J]. Biotechnology and Bioengineering, 2020,117(1): 17-29. DOI:10. 1002/bit.27169
[16]BÉJÀ O, ARAVIND L,KOONIN E V, et al. Bacterial rhodopsin:Evidence for a new type of phototrophy in the sea[J]. Science,2000, 289(5486):1902-1906. DOI:10.1126/science.289.5486.1902
[17]BROWN L S, JUNG K H. Bacteriorhodopsin-like proteins of eubacteria and fungi:The extent of conservation of the haloarchaeal proton-pumping mechanism[J]. Photochemical amp; Photobiological Sciences, 2006,5(6): 538-546. DOI:10.1039/b514537f
[18]CLAASSENS N J, VOLPERS M,SANTOS V A P M D, et al. Potential of proton-pumping rhodopsins: Engineering photosystems into microorganisms[J]. Trends in Biotechnology, 2013,31(11):633-642. DOI:10.1016/j.tibtech.2013.08.006
[19]ENGQVIST M K M, MCLSAAC R S,DOLLINGER P, et al. Directed evolution of Gloeobacter violaceus rhodopsin spectral properties[J]. Journal of Molecular Biology, 2015,427(1), 205-220. DOI:10.1016/j.jmb.2014.06.015
[20]COOLEY J W, TUKEY J W. An algorithm for the machine calculation of complex Fourier series[J]. Mathematics of Computation, 1965, 19(90):297-301.
[21]SHUICHI K, PITOR P,MARIA P, et al. AAindex:Amino acid index database, progress report 2008[J]. Nucleic Acids Research, 2008, 36(Database):D202-D205. DOI:10.1093/nar/gkm998
[22]BENSON D C. Digital signal processing methods for biosequence comparison[J]. Nucleic Acids Research, 1990, 18(10):3001-3006. DOI:10.1093/nar/18.10.3001
[23]YANG K K, WU Z,BEDBROOK C N, et al. Learned protein embeddings for machine learning[J]. Bioinformatics,2018, 34(15):2642-2648. DOI:10. 1093/bioinformatics/bty178
[24]NWANKWO N, SEKER H. Digital signal processing techniques: Calculating biological functionalities[J]. Journal of Proteomics amp; Bioinformatics, 2011,4(12):260-268. DOI:10. 4172/jpb.1000199
[25]LEVITT M. A simplified representation of protein conformations for rapid simulation of protein folding[J]. Journal of Molecular Biology, 1976,104(1): 59-107. DOI:10.1016/0022-2836(76)90004-8
[26] CEDANO J,ALOY P,PÉREZ-PONS J A,et al.Relation between amino acid composition and cellular location of proteins[J]. Journal of Molecular Biology,1997,266(3):594-600. DOI:10.1006/jmbi.1996.0804.
[27]FINKELSTEIN A V, BADRETDINOV A Y,PTITSYN O B. Physical reasons for secondary structure stability:Alpha-helices in short peptides [J]. Proteins,1991, 10(4):287-99.DOI:10.1002/prot.340100403.
A machine learning study on gloeobacter violaceus rhodopsin spectral properties
JIA Lili, SUN Tingting
(School of Sciences College,Zhejiang University of Science and Technology,Hangzhou310023,China)
In recent years, artificial intelligence technologies such as machine learning have been applied to protein engineering, and have shown unique advantages in studies on as protein structure, function prediction, and catalytic activity. In the absence of protein structure, combining protein sequence and functional properties with machine learning is a new research direction. In this papers, based on a new sequence-activity relationship (ISAR) method, the mutant library of gloeobacter violaceus rhodopsin (GR) and the maximum absorption wavelength of the spectrum are modeled by machine learning. It can fit the best model even in the case of a small number of data sets. The proposed method digitizes the protein amino acid sequence, preprocesses it through fast Fourier transform (FFT), and then performs partial least squares regression (PLSR) modeling. Finally, the best model of the amino acid sequence of the rhodopsin mutant protein and the maximum absorption wavelength of the spectrum is obtained. Modeling with the best index LEVM760106, the coefficient of determination is thatR2is 0.944, and the minimum mean square errorEis 11.64. In contrast, when the wavelet transform was used to preprocess the data, the coefficient of determination is close to 0.944, but theEis greater than 11.64, not as good as the result of FFT preprocessing. It is shown that, this method effectively solves the mathematical model relationship between protein sequence and functional characteristics, and provides support for predicting better mutants in later protein engineering.
machine learning; digital signal processing (DSP); spectral characteristics
Q 332
A
1008⁃9497(2022)03⁃280⁃07
10.3785/j.issn.1008-9497.2022.03.003
2021⁃03⁃02.
浙江省自然科学基金资助项目(LY17A040001).
郏丽丽(1993—),ORCID:https://orcid.org/0000-0002-3215-5627,女,硕士,主要从事机器学习、生物统计研究.
通信作者,ORCID:https://orcid.org/0000-0003-1388-3458,E-mail:tingtingsun@zust.edu.cn.
猜你喜欢
作物学报(2022年2期)2022-11-06
北京航空航天大学学报(2022年8期)2022-08-31
农业工程学报(2022年8期)2022-08-08
中国畜牧杂志(2022年6期)2022-06-13
作物学报(2022年8期)2022-05-29
作物学报(2022年6期)2022-04-08
黑龙江大学自然科学学报(2022年1期)2022-03-29
中国食品(2020年13期)2020-07-29
恋爱婚姻家庭·养生版(2018年3期)2018-03-24
