
聚乳酸/聚丁内酰胺电纺核-壳结构纤维的制备及性能
2022-05-25 06:07张媛婷赵黎明邱永隽
功能高分子学报 2022年2期
张媛婷, 明 远, 陈 涛,3, 赵黎明, 邱永隽
(华东理工大学 1.材料科学与工程学院, 上海市先进聚合物材料重点实验室; 2.生物工程学院, 生物反应器工程国家重点实验室, 发酵工业分离提取技术研发中心;3.中国轻工业生物基材料工程重点实验室, 上海 200237)
核-壳结构纤维的制备与应用在近年来的研究中得到了广泛关注[1]。目前,通过静电纺丝直接获得核-壳结构纤维的方法主要有同轴电纺、乳液电纺和不相容聚合物的共混电纺,其中,同轴电纺的应用最为广泛。Alharbi等[2]通过同轴静电纺丝,制备了可用于组织工程的聚乳酸/聚乙烯醇核-壳结构纤维,提高了纤维的力学性能和细胞相容性。同轴电纺需要使用具有特殊结构的同轴喷丝头,并需严格控制溶液浓度及纺丝工艺参数,以确保在喷丝头处形成核层溶液被壳层溶液完全包覆的复合泰勒锥[3],装置和工艺较为复杂。乳液电纺是通过单喷丝头从非均相的乳液中制备核-壳结构纤维的方法,装置相对简单。Su等[4]通过乳液电纺制备了聚己内酯/聚氧化乙烯核-壳结构纤维,有望应用于伤口敷料,但在乳液配制过程中,需要加入表面活性剂以保证乳液体系的稳定。一些热力学不相容的聚合物共混体系能够在静电纺丝过程中发生自发的相分离,使纤维形成核-壳结构。Tipduangta等[5]将亲水的聚乙烯吡咯烷酮与疏水的琥珀酸羟丙基甲基纤维素共混纺丝,得到了具有核-壳结构的共混纤维,并将其应用于药物的控制释放,结果表明除了聚合物相容性等热力学因素外,分子运动和溶剂挥发等动力学因素也会影响核-壳结构的形成。通过共混纺丝制备核-壳结构纤维,能够简化纺丝工艺,且避免在体系中引入其他成分。
聚乳酸(PLA)是一种生物基来源并可完全生物降解的脂肪族聚酯。由于具有良好的生物相容性和生物可吸收性,PLA特别是聚L-乳酸(PLLA)已经广泛应用于生物医用领域,同时因其良好的可加工性和环境友好的生物降解性,PLA有望在工业生产和日常生活中发挥更大作用[6,7]。PLLA的疏水性及较大的脆性限制了其进一步应用,因此在应用中常通过嵌段共聚[8],或与其他材料共混[9]等方法改善制品的性能。聚丁内酰胺(PBL,又称聚酰胺4、尼龙4)是一种结构单元中含有4个碳原子的尼龙,具有良好的力学性能,亲水性接近于棉、蚕丝等天然纤维[10]。PBL的原料γ-氨基丁酸(GABA)可以通过生物发酵法生产[11],因而属于生物基尼龙。此外,相比其他尼龙,PBL生物降解性良好,在海水、土壤中及生物体内均可发生降解[12-14]。将PBL与PLLA复合有望利用PBL柔韧、亲水的特性,在改善PLLA性能的同时不丧失材料的生物基特质和可生物降解性。Kim等[15]制备的PLLA与PBL嵌段共聚物具有生物降解性,各嵌段发生微相分离并各自结晶,但嵌段共聚的过程仍然相对复杂。本课题组在前期研究中观察到,将PLLA与PBL共混纺丝可能会形成类似于核-壳的结构,但尚未对PLLA/PBL共混纤维的性能以及具体的相分离结构进行系统的的表征和分析[16]。
本文以六氟异丙醇(HFIP)为共溶剂溶解PLLA与PBL,通过静电纺丝制备了全生物基可降解PLLA/PBL共混纤维,系统地探究了共混比例对纤维形貌、直径、亲水性、热性能和结晶性能的影响,并使用多种方法对纤维的内部结构进行了分析。研究表明共混纤维中的PLLA与PBL在单喷头电纺的过程中发生相分离,形成了以PLLA为壳、PBL为核的核-壳结构,为核-壳结构纤维的制备及其工艺的简化提供了新思路。
1 实验部分
1.1 原料和试剂
L-丙交酯(L-LA):纯度98%,梯希爱(上海)化成工业发展有限公司;PLLA:L-LA开环聚合制得,黏均分子量(Mη)为 3×104;α-吡咯烷酮(Py):纯度 99%,恒天生物基材料工程技术(宁波)有限公司;PBL:Py 开环聚合制得[16],Mη=2×104;HFIP:纯度 99.5%,上海迈瑞尔化学技术有限公司;二氯甲烷(DCM):分析纯,上海泰坦科技有限公司。
1.2 测试与表征
扫描电子显微镜(SEM):日本Hitachi 公司S-4800型,在观察前先对样品表面进行喷金。
高分辨透射电子显微镜(TEM):日本Hitachi 公司JEM-2100型,将纤维直接收集在铜网上,使用TEM观察纤维内部结构。
差示扫描量热(DSC)仪:美国PerkinElmer公司DIAMOND DSC型,先升温至190 ℃消除热历史,再以10℃/min的速率降至室温,最后再以10 ℃/min的速率升至190 ℃,实验在N2气氛下进行,N2流速为20 mL/min。
X射线衍射(XRD)仪:日本Rigaku株式会社18kW/D/max2550VB/PC 型,使用 Cu-Kα 辐射管(λ=0.154 nm),在室温下进行广角 X 射线衍射(WAXD)分析,设置电压为 40 kV,电流为 100 mA,扫描范围为 5°~55°。
接触角测量仪:上海中晨数字技术设备有限公司JC200D3型,测量去离子水在纤维膜表面的接触角。
傅里叶变换红外光谱(FT-IR)仪:美国赛默飞世尔科技公司Nicolet™ iS50型,使用FT-IR的衰减全反射(ATR)模式直接分析纤维膜的化学组成。
1.3 实验步骤
1.3.1 PLLA/PBL共混电纺纤维膜的制备 以HFIP为溶剂,PLLA、PBL或其共混物为溶质配制纺丝溶液,在室温下磁力搅拌至溶质完全溶解、溶液澄清透明。将溶液在纺丝前超声振荡30 min后,再静置30 min排出气泡,吸入注射器中并安装于微量注射泵(保定申辰泵业有限公司SPLab01-E型)上,用直流高压电源(大连泰思曼科技有限公司TE-4020P50-50型)提供纺丝电压,在电压30 kV、溶液流速1.5 mL/h、接收距离12 cm的条件下进行溶液静电纺丝,在低速旋转(200 r/min)的辊筒收集装置上收集静电纺丝纤维,约30 min后,从辊筒上取下由纤维堆积而成的纤维膜(固定溶液中聚合物的质量分数为2.0%,当聚合物中PBL的质量分数(w(PBL))分别为0,30%、50%、70%、80%、100% 时,所得纤维膜分别简称为B-0、B-30、B-50、B-70、B-80、B-100)。
1.3.2 PLLA/PBL共混纤维膜溶剂刻蚀实验 将PLLA/PBL共混纤维膜在室温下真空干燥并称重,浸泡在装有20 mL DCM的样品瓶中,定时取出纤维膜,用DCM冲洗后,室温下真空干燥并称重,用ATR分析纤维膜成分,用SEM观察纤维形貌。将称量后的纤维膜放入装有新DCM的样品瓶中浸泡,重复上述操作。
2 结果与讨论
2.1 PLLA/PBL共混纤维的形貌与直径
图1 所示为PLLA/PBL单组分及共混纤维膜的SEM图像。不同共混比例的溶液均可得到无串珠或带状结构,外表光滑的纤维。纯PLLA纤维直径分布比较均匀,而加入PBL共混纺丝得到的纤维中都同时出现了一定数量的直径约0.1 μm的细纤维和直径在1.0 μm以上的粗纤维。在此前的报道[5]中,其成因主要为溶液黏度较大,喷丝头发生部分堵塞而产生了多泰勒锥,或电纺过程中溶液射流劈裂形成了“小二次射流”。溶液受到较高的外加电压或溶液本身较高的电导率引起射流表面电荷不稳定[17],溶液射流中不均匀的电荷分布导致部分区域的静电斥力克服了表面张力,以及不均匀的分子量分布造成部分区域黏度较低[18],以上现象均可能导致射流的劈裂。此外,共混溶液的相分离也是纤维直径分布不均匀的一个原因[19]。本实验中PBL的加入导致溶液电导率升高,体系中PLLA、PBL两种组分引起的局部黏度不均匀性,以及在纤维形成过程中发生的聚合物相分离是造成共混纤维中产生较多细纤维的可能原因。

图1 PLLA/PBL共混纤维的SEM图像Fig.1 SEM images of PLLA/PBL blend fibers
图2所示为PLLA/PBL纤维平均直径随w(PBL)的变化情况。由图可知,纯PLLA纤维的平均直径较大,为0.857 μm,加入PBL后,由于PBL的极性较强,在电纺时表现出聚电解质效应,使溶液导电性增强,纤维平均直径随w(PBL)的增加而显著减小[20]。
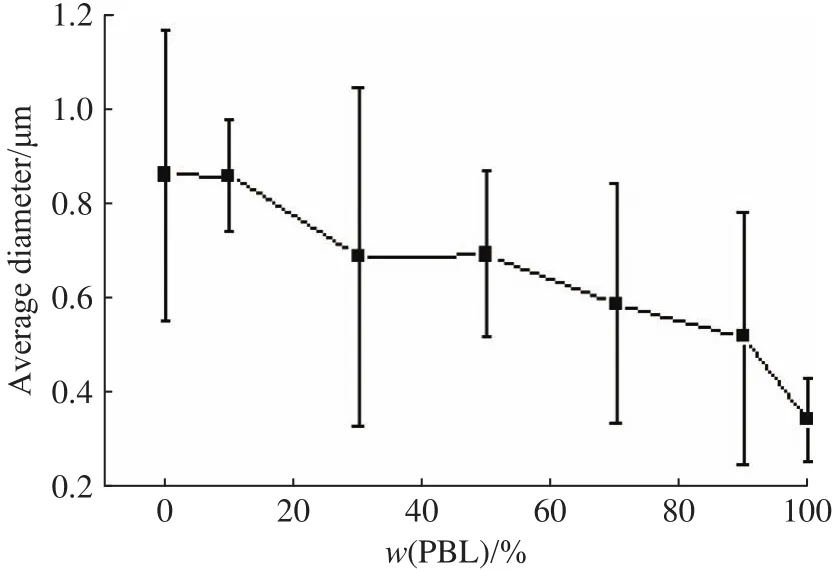
图2 纤维平均直径随PBL质量分数的变化Fig.2 Fiber average diameters versus PBL mass fractions
2.2 PLLA/PBL共混纤维的热性能与结晶性能
2.2.1 纤维中PLLA结晶的DSC分析 由于PBL的熔融温度与热分解温度接近(约275 ℃[20]),为避免PBL的分解,仅使用DSC对纤维中PLLA组分的热性能及结晶性能进行分析。PLLA/PBL共混纤维消除热历史后降温(图3(a))及二次升温(图3(b))过程的DSC曲线如图3所示。
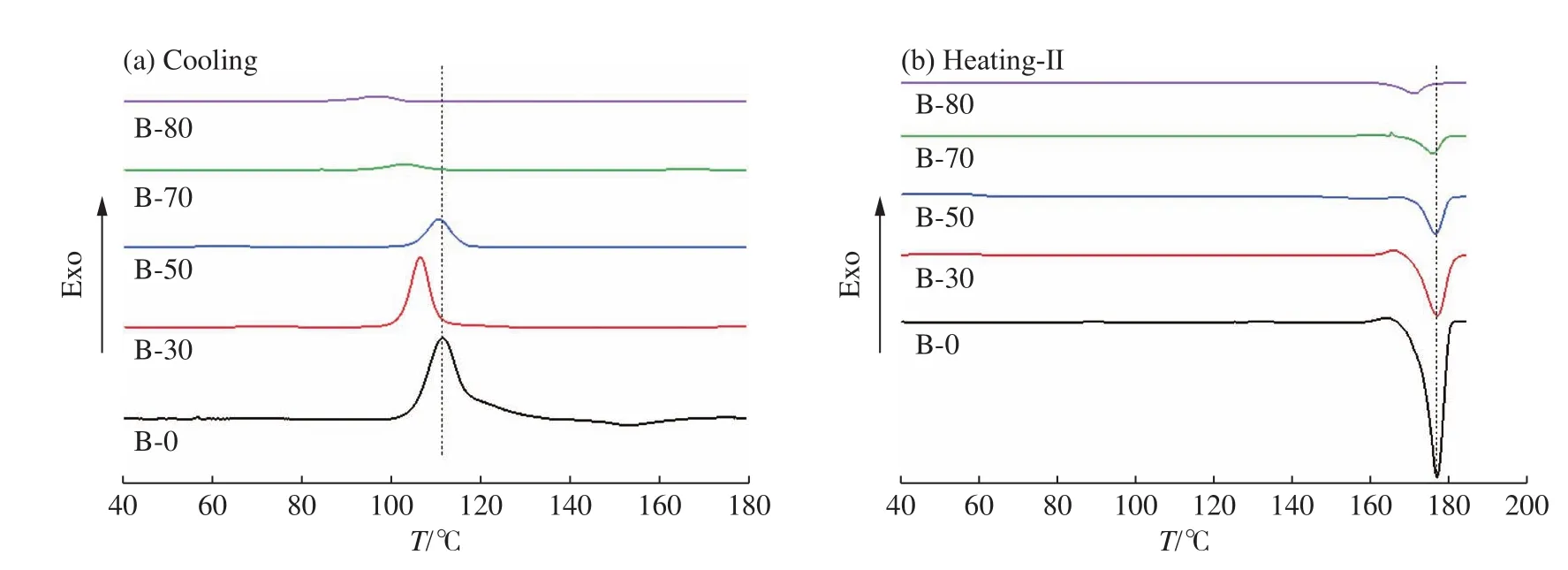
图3 PLLA/PBL共混纤维的DSC曲线Fig.3 DSC curves of PLLA/PBL blend fibers
在降温曲线上能够观察到PLLA的熔体结晶峰(90~115 ℃),而在二次升温曲线上能够观察到PLLA的熔融峰(170~180 ℃)。与纯PLLA纤维相比,PLLA/PBL共混纤维熔体结晶温度与二次升温的熔融温度都有所降低,其中熔体结晶温度的变化更为明显,特别是B-80的熔体结晶温度较纯PLLA纤维降低了约15 ℃,该样品在二次升温时的熔融温度也较纯PLLA纤维降低了约6 ℃。
由于PBL的熔融温度远高于PLLA的熔融温度,因此共混纤维在加热至PLLA熔融温度以上时,纤维中的PBL仍然保持原来结构,并对PLLA的熔体结晶与熔融产生影响。据报道,共混电纺纤维中或同轴电纺纤维核层的低熔融温度组分在熔体结晶过程中会受到尚未(或未完全)熔融的高熔融温度组分产生的两种相互竞争的效应:界面诱导效应和纳米约束效应[21,22]。界面诱导效应即高熔融温度的组分在相分离的界面处起到成核剂的作用,诱导和加速低熔融温度组分的结晶。纳米约束效应则是由于纤维的径向尺寸有限,使得聚合物在相分离后形成更小的微区,这些微区中缺少能够作为晶核的微晶,同时微区的尺寸会限制晶体生长,因此结晶过程受到了约束,需要更高的过冷度才能结晶。
对于样品B-70和B-80,结晶温度和熔融温度的明显降低可能是由于大量的PBL倾向于形成连续相,PLLA结晶受到了较强的约束效应使得结晶温度显著降低,而其熔融温度的降低也反映了降温过程中形成的晶体完善程度较低。对于样品B-50,在相分离时可能会在纤维中形成两相互穿程度较高的结构,从而使得两相界面增大,加强了界面诱导成核的作用,其结晶温度和熔融温度与纯PLLA纤维相比降低较少。
2.2.2 初纺与退火纤维的XRD分析 初纺纤维的XRD图像如图4 (a)所示,图中仅能观察到21.1°和24.0°处的晶体衍射峰,分别对应于PBL的α-晶型的(2 0 0)和(0 2 0)晶面,没有观察到与PLLA有关的晶体衍射峰,说明初纺纤维中PLLA组分的结晶程度较低,这是由于静电纺丝过程中射流受到的高速牵伸作用以及溶剂的快速挥发,使得离开喷丝头的溶液射流在飞行过程中快速固化,部分分子链没有来得及形成晶体结构,与文献[23]中报道的PLLA初纺纤维结晶情况一致。
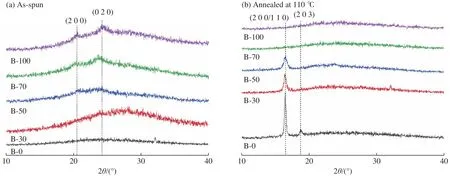
图4 PLLA/PBL共混纤维膜的XRD图像Fig.4 XRD patterns of PLLA/PBL blend fiber membranes
纤维在110 ℃退火后的XRD图像如图4 (b)所示,图中16.4°和18.7°处出现的尖锐晶体衍射峰对应于PLLA的α-晶型的(2 0 0/1 1 0)和(2 0 3)晶面。w(PBL)增大后,退火后的α-晶体衍射峰强度明显降低,半峰宽变宽,说明结晶度和晶体完善程度降低,该现象在PLLA与聚乙醇酸共混纺丝实验中也有过报道[24]。
2.3 PLLA/PBL共混纤维膜表面的亲、疏水性
PLLA/PBL共混纤维膜表面的接触角和水滴形态如图5 所示。纯PLLA纤维膜表面呈疏水性,纯PBL纤维膜表面呈亲水性,而PLLA/PBL共混纤维膜表面的水接触角虽然随w(PBL)的增大而略有减小,但始终处于疏水状态。由此推测,纤维表面的主要成分可能是疏水的PLLA,PBL的加入对于纤维膜表面的亲、疏水性影响不大,说明该组分可能主要存在于纤维内部。

图5 PLLA/PBL共混纤维膜表面接触角及水滴形态Fig.5 Contact angles and droplet morphologies on PLLA/PBL blend fiber membrane surfaces
2.4 PLLA/PBL共混纤维的内部结构
2.4.1 TEM 为了更加直观地了解共混纤维的内部结构,使用TEM观察了直接收集在铜网上的纤维,B-50样品中直径较粗与直径较细的纤维的TEM图像如图6所示。在TEM图像中观察到,直径较粗的纤维中形成了衬度不同的两层,内层较暗,外层较亮,两层之间有比较清晰的界面,与文献[25, 26]报道的核-壳结构类似,而直径较细的纤维中没有观察到此类结构。

图6 PLLA/PBL共混纤维膜的TEM图像(w(PBL) = 50%)Fig.6 TEM images of PLLA/PBL blend fiber membrane (w(PBL)= 50%)
2.4.2 溶剂刻蚀实验w(PBL) 为30%、50%、70%的PLLA/PBL共混纤维膜经溶剂刻蚀后仍然保持着圆柱状的纤维形貌(图7),平均直径较刻蚀之前都有所减小,分别为原来的59.9%,69.0%,95.6%。DCM对PBL轻微的溶胀作用导致刻蚀后纤维直径占原直径的比与w(PBL)存在偏差。右上角放大的纤维图像中观察到刻蚀后的纤维表面出现了一些沟壑,可能是纤维去除PLLA组分后在干燥过程中形成的。此外,溶剂刻蚀后纤维表面没有出现孔洞结构,说明纤维内部基本没有PLLA组分存在。

图7 PLLA/PBL共混纤维膜DCM刻蚀后的SEM图像Fig.7 SEM images of PLLA/PBL blend fiber membranes etched by DCM
PLLA/PBL共混纤维膜的质量随DCM刻蚀时间的变化如图8 (a)所示,其中纵坐标为剩余质量百分比(即刻蚀后样品质量占初始质量的百分比)。由图可见,在刻蚀前15 min,所有样品质量迅速减小,随后基本趋于稳定,剩余质量占初始质量的百分比与w(PBL)接近,其中样品B-50剩余质量百分比略高于50%。这一结果也表明PLLA大多处于纤维外层,可迅速溶解在DCM中,从而造成纤维质量快速减小。

图8 PLLA/PBL共混纤维膜的(a)剩余质量随刻蚀时间的变化情况以及(b~d)在不同刻蚀时间的ATR谱图Fig.8 (a) PLLA/PBL fiber membrane residual mass versus etching time and (b—d) ATR spectra at different etching time
图8 (b~d)所示为PLLA/PBL共混纤维膜在DCM中刻蚀不同时间的ATR谱图。由于ATR分析中,光线穿透样品的深度为1~4 μm,超过了大多数纤维的直径,因此认为纤维膜的ATR谱图反映的是纤维整体的化学组成。谱图中波数在1 757 cm−1和1 089 cm−1处的峰分别为PLLA分子中C=O和C-O-C 伸缩振动峰,反映了样品中PLLA组分的存在及含量变化。样品B-30和B-70在刻蚀15 min后几乎检测不到PLLA特征基团峰,说明纤维中的PLLA在短时间内已经完全被DCM溶解;样品B-50的PLLA特征基团峰在刻蚀15 min后峰面积明显减小,但刻蚀24 h后仍可在1 757 cm−1处观察到较小的峰,说明该样品中PLLA的溶解较慢,也验证了前述纤维在两相界面形成互穿结构的推论。刻蚀后的纤维中酰胺Ⅰ(约1 635 cm−1)、酰胺Ⅱ(约1 540 cm−1)等PBL特征基团峰仍然存在,且没有明显变化,说明此时纤维剩余成分主要为PBL,且DCM的刻蚀对PBL影响较小。溶剂刻蚀后直径较细的纤维依然存在,形貌上仅发生了轻微溶胀,说明它们是由PBL 单独形成的。
上述溶剂刻蚀实验的结果充分表明,PLLA/PBL共混纤维中PLLA主要分布在壳层,而 PBL形成连续的纤维芯层,这些结果与TEM图像中显示的核-壳结构相符合。该结构的形成可能是由于PBL分子间氢键作用力强,在溶液中分子链缠结程度高,运动能力弱,因此在相分离时留在了纤维的中心形成核层,而PLLA分子运动能力强,会在纺丝过程中向溶液射流的外侧迁移形成壳层。
根据聚合物溶液共混的相关理论,在分子量一定的情况下,当两种聚合物质量分数相差较大时,随着溶剂的挥发,系统组成在相图上的路径只与双节线相交,聚合物按照成核-生长机理发生相分离,倾向于形成核-壳结构。而当两种聚合物质量分数接近时,随着溶剂的挥发,系统组成在相图上的路径只与旋节线相交,或先与双节线相交再与旋节线相交,两种聚合物部分或全部按照旋节相分离机理发生相分离,倾向于形成互穿结构,或者形成在两相界面处互穿程度较高的核-壳结构[27]。样品B-50在溶剂刻蚀时PLLA组分溶解较慢,刻蚀后纤维表面的沟壑最为明显,可能是因为PLLA与PBL含量接近。尽管PLLA同样存在于纤维外层,但与内层PBL互穿程度较高,PLLA的溶出受到了阻碍,这与2.2.1节中得到的推论相印证。
综上所述,通常情况下需要采用同轴电纺等方法才能获得的核-壳结构,PLLA与PBL通过简单的溶液共混及单喷头电纺即可形成,该简化方法为制备核-壳结构纤维提供了新思路。
3 结 论
(1)固定纺丝溶液中聚合物的质量分数为2.0%,PLLA/PBL溶液通过静电纺丝可得到表面光洁、均匀无珠的纤维,直径在0.1~1.6 μm。PLLA/PBL共混纤维的平均直径随PBL含量的增加而减小,且直径分布较宽。
(2)初纺纤维中PLLA组分的结晶度较低,在退火或熔融降温后PLLA会发生结晶。共混纤维中PBL相区的存在会抑制PLLA的熔体结晶和110 ℃退火时的结晶,降低结晶度和晶体的完善程度。
(3)电纺过程中两种聚合物发生了相分离,形成了PLLA在外层、PBL在内核的核-壳结构,PBL的引入对改善PLLA/PBL共混纤维膜表面的亲水性效果不明显。
猜你喜欢
学与玩(2022年12期)2023-01-11
九江学院学报(自然科学版)(2022年2期)2022-07-02
陶瓷学报(2020年2期)2020-10-27
中国特种设备安全(2019年2期)2019-04-22
燕山大学学报(2015年4期)2015-12-25
中国塑料(2015年6期)2015-11-13
中国塑料(2015年7期)2015-10-14
装备机械(2015年1期)2015-02-28
火炸药学报(2014年1期)2014-03-20
中成药(2014年9期)2014-02-28
