
Castleman病变异型POEMS综合征
2022-05-18 02:28周玉超许书添李世军
肾脏病与透析肾移植杂志 2022年2期
周玉超 许书添 李世军
病例摘要
30岁男性,因“水肿、蛋白尿2月余,腹胀、血清肌酐(SCr)升高1月余”于2020-08-12收入院。
现病史患者2020年5月下旬于劳累后出现眼睑、双下肢水肿,6月1日就诊于当地人民医院,查血压高 161/100 mmHg,尿蛋白定量1 768 mg/24h,白蛋白33.4 g/L,SCr 67.5 μmol/L,血常规未见异常。当地行肾活检病理诊断为“血栓性微血管病样病变”,给予缬沙坦氨氯地平片85 mg,2次/d及百令胶囊、肾炎康复片治疗。7月初患者出现腹胀不适,食欲明显下降。7月20日因腹胀症状进行性加重再次住院,复查尿蛋白定量553 mg/24h,血清白蛋白降低至24~29 g/L,SCr升至130 μmol/L,血常规未见异常,胸部CT示“双侧胸腔积液、心包积液、腹腔积液”,超声示“肝脏体积增大,脾脏体积稍增大”。予腹腔穿刺置管引流腹水,查腹水有核细胞计数265个/μl,蛋白总量24.47 g/L,腹水抗酸染色、培养、腺苷脱氨酶、结核杆菌DNA等病原学检测均为阴性。引流腹水后患者腹胀症状有所减轻。8月11日出现左上腹痛,转诊至东部战区总医院急诊科,体温正常,SCr 150 μmol/L,血钾5.6 mmol/L,降钙素原 3.97 μg/L,C反应蛋白6.1 mg/L,给予莫西沙星0.4 g/d抗感染、止痛、降钾等治疗,为进一步诊治收住院。患者病程中精神欠佳,体力、食欲明显下降,体重下降约5 kg,大便正常,尿量有所减少(400~1 000 ml/d)。无发热、皮疹、关节痛、腹泻、呕吐、黄疸等表现。
既往史无特殊。
个人史吸烟10~20支/d十余年,偶尔饮酒,从事木工工作,余无特殊。
婚育史、家族史无特殊。
体格检查体温36℃,脉搏82次/min,呼吸15次/min,血压 139/97 mmHg,皮肤偏黑,指甲发白(图1),双侧颈部、右侧腋窝和双侧腹股沟可触及肿大淋巴结,质中,活动度良好,无压痛。两下肺呼吸音减低,无干湿啰音,腹部明显膨隆,腹壁静脉未见曲张,反麦氏点可见一腹腔引流管在位,引流出淡黄色清亮液体,移动性浊音(+),肝脾肋下未触及,双下肢轻度水肿。痛觉过敏,四肢肌力、肌张力正常,脑膜刺激征阴性,双侧病理征阴性。
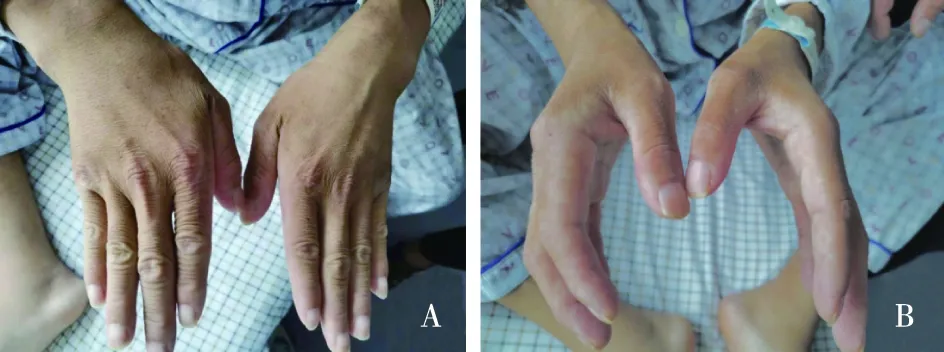
图1 A:患者双手手背肤色偏黑;B:患者手背与手掌肤色差异明显
实验室检查
尿液 蛋白±,隐血阴性,蛋白定量0.32 g/24h,C3 2 mg/L,α2-MG 2 mg/L,NAG 13.8 U/(g·Cr),RBP 1 mg/L,尿本周蛋白阴性。
血常规 白细胞计数12.74×109/L,血红蛋白155 g/L,血小板417×109/L,C反应蛋白 6.1 mg/L。
凝血功能 PT 13.6 s,INR 1.19,APTT 35 s,TT 16 s,Fbg 4.49 g/L。
血生化 降钙素原3.97 μg/L,白细胞介素6(IL-6)26.86 ng/L,脑利钠肽前体 45.18 pmol/L,白蛋白26.5 g/L,球蛋白 28.6 g/L,谷丙转氨酶11 U/L,谷草转氨酶13 U/L,胆红素正常,尿素19.2 mmol/L,SCr 150 μmol/L,尿酸 511 μmol/L,血钾 5.6 mmol/L,血钠 136 mmol/L,血氯 107 mmol/L,总二氧化碳 16.4 mmol/L,乳酸脱氢酶 390 U/L,胆碱酯酶 1.7 KU/L,肌钙蛋白、肌红蛋白、肌酸激酶、肌酸激酶-MB无升高;叶酸 3.65 ng/ml,维生素B12 49 pmol/L。
免疫学检查 ANA 1∶ 128,A-dsDNA阴性,补体C3 0.983 g/L,C4 0.277 g/L。抗磷脂酶A2受体抗体 2.99 RU/ml。体液免疫:IgG 10.6 g/L,IgA 1.91 g/L,IgM 0.864 g/L,血液单特异性游离轻链:κ 105 mg/L,λ 172 mg/L,κ/λ 0.61,复查:κ 165 mg/L,λ 172 mg/L,κ/λ 0.96,免疫固定电泳未见异常单克隆免疫球蛋白条带。
内分泌检查 甲状腺功能:T3 0.41 nmol/L(参考值范围1.01~2.48 nmol/L),FT3 2.5 pmol/L(参考值范围 3.8~6.5 pmol/L),TSH 6.16 mIU/L(参考值范围 0.49~4.5 mIU/L);性激素:睾酮 1.81 nmol/L(参考值范围 9.4~37 nmol/L),脱氢表雄酮 1.5 μmol/L(参考值范围 2.31~12.59 μmol/L),催乳素 538.4 mIU/L(参考值范围 95.4~400 mIU/L)。
其他腹水常规及生化:淡黄色,微混浊,李凡它实验阳性,白细胞计数120个/μL,红细胞计数 400个/μL,总蛋白 25.4 g/L,乳酸脱氢酶 334 U/L,葡萄糖 4.5 mmol/L,氯 114 mmol/L;腹水培养:无菌生长。传染病四项:均阴性。
辅助检查
心电图 窦性心律,V1-V3导联ST段略呈弓背型,电轴左偏。
超声 双肾:左肾121 mm×57 mm×60 mm,右肾120 mm×50 mm×60 mm,肾皮质回声增强,结构不清楚。
心脏:中~大量心包积液。
消化系统:肝静脉、门静脉超声未见异常,下腔静脉超声示近心段内径4 mm,肝段内径5.2 mm,提示下腔静脉内径偏细。
浅表超声:双侧颈部可见肿大淋巴结,较大的约21 mm×8 mm(右侧)、26 mm×8 mm(左侧),右侧腹股沟见肿大淋巴结(较大的约13 mm×4 mm)。
CT 两肺散在炎症,双侧胸腔积液伴两下肺部分萎陷,心包积液,纵隔及双侧腋窝多发略大淋巴结,腹盆腔大量积液引流后改变,腹盆壁软组织水肿。
下腔静脉CTV 下腔静脉肝段管腔狭窄,管腔内未见充盈缺损,同时可见肝脾肿大、腹膜后多发肿大淋巴结、腹盆腔大量积液、腹盆壁软组织水肿。
PET-CT 双侧颈部、左侧锁骨区、双侧腋窝、纵膈、腹膜后多发淋巴结,部分FDG代谢轻度增高(淋巴结增生?),左侧髂骨病变,局部FDG代谢增高(浆细胞浸润?),双侧胸腔、心包、腹盆腔积液,腹盆壁皮下水肿,肝脾轻度肿大。
骨髓检查 (右侧髂前上棘)骨髓细胞学未见异常,(左侧髂前上棘)骨髓流式细胞学:未检出单克隆浆细胞。
眼底检查 未见视乳头水肿。
肾脏病理外院肾活检光镜切片经国家肾脏疾病临床医学研究中心肾脏病理医师重新阅片。肾小球毛细血管袢开放欠佳,内皮细胞肿胀,节段内皮增生致袢腔狭窄、闭锁,袢腔内未见微血栓,系膜区增宽,系膜基质疏松,内皮下间隙增宽,节段双轨形成,间质小动脉及弓状动脉内膜轻度黏液样变性,管壁增厚,管腔狭窄,节段动脉壁可见玻璃样变(图2);当地医院免疫荧光:IgM±,余均阴性;电镜示肾小球毛细血管袢基膜内皮下间隙明显增宽,其内可见絮状物,偶见系膜基质插入基膜内皮下,未见确切电子致密物沉积。该患者主要病变在肾小球,突出特点为内皮细胞增生、内皮下疏松、系膜溶解等改变。间质血管有内膜增厚、管壁透明样变性等病变。
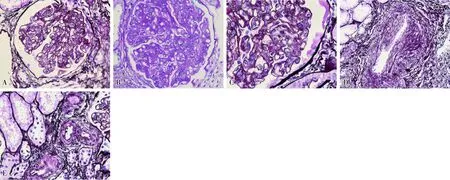
图2 A、B:肾小球内皮细胞肿胀,节段内皮增生,系膜区增宽,系膜基质疏松(A:PASM,×400;B:PAS,×400);C:肾小球内皮下间隙增宽,节段双轨形成(PASM,×600);D:间质小动脉管壁增厚(PASM,×400);E:间质小动脉内膜增厚、管壁透明样变性(PASM,×400)
诊疗分析
总结本病例最主要的特点包括:肝脾肿大和多发淋巴结肿大、多浆膜腔积液(胸腔积液、腹腔积液、心包积液)及皮肤软组织水肿提示血管外容量超负荷、肾小球内皮病变及系膜溶解,诊断考虑如下。
血栓性微血管病患者于外院病理诊断为“血栓性微血管病样病变”,但无贫血、血小板减少,血红蛋白及血小板计数水平反而偏高,不符合溶血尿毒综合征(HUS)和血栓性血小板减少性紫癜(TTP)等典型TMA相关疾病的临床特点。经病理会诊,本例符合系膜溶解、内皮下疏松病理特点,微血管病变不典型。
POEMS综合征POEMS综合征为一组伴随浆细胞肿瘤的临床综合征,其临床表现常包括:多发性周围神经病变(polyneuropathy),脏器肿大(Organomegaly),内分泌疾病(endocrinopathy),单克隆免疫球蛋白(M-protein),皮肤改变 (skin changes)。但POEMS综合征的临床表现往往不限于上述5条,且这些临床表现并不一定同时呈现。根据2019年关于POEMS综合征诊断进展的最新文献[2],其诊断标准如表1。

表1 POEMS综合征诊断标准[2]
(1)符合两条强制性诊断标准,至少1条主要诊断标准,至少1条次要诊断标准,可诊断POEMS综合征;(2)Castleman病变异型POEMS综合征可无单克隆浆细胞增殖的相关证据
本例有器官肿大、血管外容量超负荷(胸腔积液、腹腔积液、心包积液、皮肤软组织水肿)、内分泌异常、皮肤改变以及血小板和红细胞增多等表现,符合POEMS综合征多条次要标准,但还需满足两条强制标准及至少1条主要标准才能确诊。
Castleman病Castleman病是一个病理诊断名词,也称淋巴结增生症、血管滤泡性淋巴结增生,是一组异质性反应性淋巴组织增生性疾病的总称。该组疾病具有共同的淋巴组织病理特征,但病因及临床表现多样——可表现为无症状单发淋巴结肿大,亦可表现为多系统损害(自身免疫性疾病、甚至典型POEMS综合征)。根据Castleman病协作组织(The Castleman Disease Collaborative Network,CDCN)提出的最新标准,根据病变淋巴结的分布区域可分为单中心型(UCD)和多中心型(MCD),MCD又可根据病因学分为HHV8相关性MCD(HHV8-MCD),POEMS相关性MCD(POEMS-MCD)及特发性MCD(iMCD),iMCD根据临床表现进一步分为iMCD-TAFRO及iMCD-NOS[1]。其中POEMS-MCD是指确诊MCD的同时还满足POEMS综合征的诊断标准。
患者进一步完善神经电生理检查提示存在神经性受损(在肌电图检查过程中,患者因难以忍受疼痛无法配合完成所有检查,考虑与其皮肤痛觉敏感性增加有关)。血清血管内皮生长因子(VEGF)明显升高(1 115.49 pg/ml,参考值范围0~142 pg/ml)。因CT示腹膜后多发淋巴结肿大,浅表超声示双侧颈部、腋窝、腹股沟多发淋巴结肿大,取左侧颈部淋巴结活检,病理诊断为系统性Castleman病(浆细胞型)(图3),淋巴结免疫组化染色HHV8标记为阴性,外周血NGS未检出HHV8序列;Castleman病诊断成立。
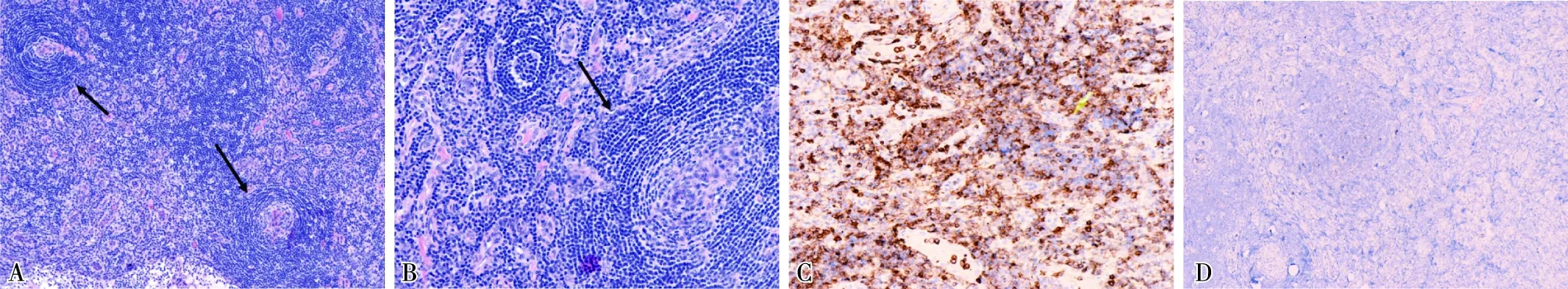
图3 A、B:淋巴结内可见“棒棒糖”、“同心圆”样淋巴滤泡(HE,×200);C:免疫组化CD20染色阳性(IH,×100);D:免疫组化HHV8染色阴性(IH,×100)
Castleman病变异型POEMS综合征本例患者多发淋巴结肿大,淋巴结活检符合CD的病理特征,符合MCD诊断标准。淋巴结免疫组化HHV8染色阴性,外周血NGS未检出HHV8,可以排除HHV8相关性MCD。尽管本病例表现出POEMS综合征的多种临床特点,包括神经病变、病理确诊的CD、VEGF水平升高、器官肿大、血管外容量超负荷、内分泌异常、皮肤改变、血小板计数及红细胞计数增多、维生素B12水平降低等。但患者无肢端麻木等症状,周围神经病变表现轻微而不典型,且未发现M蛋白及单克隆浆细胞增殖性病变,不满足POEMS综合征的强制性诊断标准,因此不能诊断POEMS-CD。回顾2011年、2019年更新的POEMS综合征诊断、风险分层及治疗相关文献,有一类相对特殊的CD患者,满足POEMS综合征的若干主要和次要标准,但外周神经损害轻微,且无单克隆性浆细胞增殖证据。此类患者应归为Castleman病变异型POEMS综合征[2-3]。
最后诊断Castleman病变异型POEMS综合征。
治疗和随访患者入院当天出现腹痛症状,且降钙素原升高,腹腔积液性质介于漏出液与渗出液之间,考虑腹腔感染。外院腹水抗酸染色、培养、腺苷脱氨酶、结核杆菌DNA等病原学检测均为阴性,予重复送检腹水常规、生化、培养,并送检病原学NGS检测。NGS报告检出铜绿假单胞菌序列,给予抗感染治疗,后患者体温持续正常,腹痛缓解,重复两次腹腔积液细菌培养无菌生长,期间监测CRP降至正常范围,但降钙素原水平持续高于正常(2.37~4.35 μg/L)。
患者病初SCr正常,随着腹胀的出现和加重,血肌酐进行性升高,肾脏B超示双肾体积略偏大,结合病程,符合急性肾损伤(AKI)。入院后给予给予呋塞米利尿,适度引流腹腔积液减轻腹胀症状,补充白蛋白改善低蛋白血症,提高血浆胶体渗透压。患者腹胀症状有所改善,腹腔积液仍持续产生,引流不尽,SCr稳定在148.5~163.5 μmd/L,但尿素氮逐渐升至28.8 mmd/L,环硅酸锆钠降钾治疗下血钾仍持续偏高(5~6 mmol/L),可能与POEMS综合征内分泌病变导致肾上腺盐皮质激素异常有关。诊断明确为Castleman病后,于8月25日加用甲泼尼龙40 mg/d,监测SCr稳定,尿素氮进一步升高至108.9 mg/dl,伴高钾血症(5.99 mmol/L),于8月27日开始连续性肾脏替代治疗(CRRT)支持治疗。9月2日起患者出现间断低热,体温最高37.5℃,复查胸部CT提示两肺炎症、两侧胸腔积液较前增多,外周血NGS报告检出嗜麦芽窄食单胞菌序列,将激素逐渐撤减至甲泼尼龙片8 mg/d,并予以莫西沙星注射剂、复方磺胺甲噁唑片抗感染,胸腔穿刺引流积液等治疗。患者降钙素原仍持续高于正常(5.3~3.7 μg/L),但体温恢复正常,持续10 d未再发热,血培养、胸腔积液培养均无菌生长,遂于9月15日加用沙利度胺片50 mg/晚。后患者因经济原因要求自动出院,出院时生命体征平稳,尿量300 ml/d,未摆脱透析。
讨 论
本文报道1例以肾脏损害为首发表现,淋巴结活检确诊为MCD,临床不完全符合POEMS综合征的病例。
POEMS综合征为一组临床综合征,从分子水平来看,VEGF被认为是与其发病机制和疾病活动联系最为紧密的细胞因子。1996年Wanatabe等[4]研究发现POEMS综合征患者血清VEGF明显升高,后续研究认为VEGF升高可作为诊断POEMS综合征的生物标志物,VEGF水平会随着病情的缓解而降低,可以用于评估疗效[5-6]。VEGF通过作用于血管内皮细胞上的VEGF受体,导致内皮细胞通透性增加,微血管渗漏,多浆膜腔积液,还可导致血神经屏障功能下降造成周围神经病变;VEGF导致肾小球内皮细胞病变,可形成血栓性微血管病样改变;VEGF促进血管生成,可导致皮肤血管瘤形成、肝脾肿大。而CD中高表达的细胞因子则主要是IL-6,在动物模型中过表达IL-6可诱发类似iMCD的相关临床表现[1],拮抗IL-6或其受体可明显改善一部分iMCD患者的症状。本例患者血清VEGF水平明显升高,多种临床表现均与VEGF生物效应吻合,而IL-6水平仅轻微升高(14.96~26.96 ng/L),VEGF与本病例的发病机制联系可能更为密切。
POEMS综合征的诊断标准指出M蛋白或单克隆浆细胞增殖为诊断POEMS综合征的两条必备条件之一。符合POEMS综合征的多种临床特征,但均未检出M蛋白和单克隆浆细胞增殖的类似情况在其他文献中亦有报道[5]。有研究认为免疫固定电泳检测M蛋白的敏感性不足可能是未能检出M蛋白的原因,有研究认为多克隆免疫球蛋白增多可导致两种游离轻链间的差异被掩盖,游离轻链比例表现为正常[7],还有病例报道M蛋白阴性,但骨髓活检发现限制性表达λ轻链的浆细胞,诊断为“非分泌性”POEMS综合征[8]。有些M蛋白阴性的不典型POEMS综合征病例在随访中发现了M蛋白[5]。在本病例中单特异性游离轻链比例、免疫固定电泳均未能发现M蛋白,骨髓涂片以及骨髓流式细胞学检测亦未能发现异常浆细胞,但VEGF已明显升高并致病,可能处于POEMS综合征的前驱阶段。
POEMS综合征累及多个系统,症状出现顺序不一,首诊科室随之不同,由于其发病率低,临床表现缺乏特异性,因此极易误诊漏诊。2020年3月北京协和医院风湿免疫科和消化内科报道一例Castleman病变异型POEMS综合征,患者以发热、胸腹腔积液、脾大为首发表现,最初考虑结核感染,抗结核治疗无效,起病3个月后方通过淋巴结活检确诊为CD。该患者具有POEMS综合征的多种表现,但M蛋白未检出,不符合POEMS强制诊断标准,最终诊断为Castleman病变异型POEMS综合征[9]。本病例以肾脏损害为首发表现。以肾脏受累为首发表现并且获得肾脏病理资料的CD病例少有报道,2012年本中心曾报道1例POEMS综合征相关肾损害病例[10]并对一组12例POEMS综合征病例进行总结分析[11]。研究指出POEMS综合征相关肾损害的主要临床表现为水肿、少量蛋白尿、SCr升高、血尿酸升高,主要病理表现为系膜溶解和内皮细胞病变,少见免疫复合物沉积,内皮损伤是导致系膜溶解的主要原因。内皮病变和系膜溶解等肾脏病理表现是诊断POEMS综合征的重要线索。
POEMS综合征预后不佳,据报道其起病至确诊平均需18月,中位生存期仅5~7年,而Castleman病伴随的POEMS综合征预后更明显差于经典的POEMS综合征[12]。上文中协和医院所报道的病例,起病至确诊历时3月,确诊后给予来那度胺联合地塞米松方案诊疗,但病情进展迅猛,错过了最佳治疗时机,最终预后不佳。本例亦历时3月方得确诊,在确诊时已出现多浆膜腔积液、急性肾损伤、感染等并发症,加之患者因经济原因中断治疗,导致预后不佳。
小结:POEMS综合征可累及多个器官和系统,临床表现多样,容易误诊漏诊,且预后不佳。本文报道1例罕见的Castleman病变异型POEMS综合征。不符合POEMS综合征全部强制性诊断标准,VEGF在其发病机制中起重要作用,肾脏病理表现为内皮病变和系膜溶解,是诊断的重要线索。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
传染病信息(2022年3期)2022-07-15
中国临床医学(2022年3期)2022-07-08
中国典型病例大全(2022年11期)2022-05-13
临床超声医学杂志(2022年4期)2022-05-07
天津医科大学学报(2021年4期)2021-08-21
医学信息(2021年5期)2021-03-21
中国生殖健康(2019年3期)2019-02-01
大众健康(2016年6期)2016-08-03
安徽医科大学学报(2015年9期)2015-12-16
