
β-烟酰胺单核苷酸跨境产品中NMN含量的测定
2022-05-14 08:22张文宇于聪聪王大宏张维冰云振宇
食品工业科技 2022年10期
张文宇,兰 韬 ,赵 溪,吴 琦,初 侨,于聪聪,王大宏,张维冰,云振宇
(1.齐齐哈尔大学,黑龙江齐齐哈尔 161006;2.中国标准化研究院,北京 100191;3.汤臣倍健股份有限公司,广东珠海 519040;4.庶正康讯(北京)商务咨询有限公司,北京 100054;5.华东理工大学,上海 200237)
β-烟酰胺单核苷酸(β-nicotinamide mononucleotide,NMN)是一种具有生物活性的核苷酸,由磷酸基和含核苷的核糖与烟酰胺反应自然形成[1],分子结构如图1所示。在动物细胞中,NMN是一种细胞能量来源,是烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD+)生物合成的代谢物,在细胞增殖和功能中起着至关重要的作用[2]。研究表明补充NMN可以提高体内NAD+含量[3],从而延缓、改善、防止衰老诱导的代谢紊乱、老年疾病等[4−9],所以NMN近年来成为生理医学领域的研究热点,并成为医药、功能性食品、化妆品等行业的珍贵原料[10−11]。由于NMN在我国还未获得药品、保健食品、食品添加剂和新食品原料许可,所以主要作为跨境产品通过跨境电商平台进入国内。

图 1 β-烟酰胺核苷酸的分子结构式Fig.1 Molecular structure formula of β-nicotinamide nucleotide
人体可自身合成少量NMN[12],也会通过摄食西兰花、卷心菜等果蔬补充NMN[13]。但由于这些果蔬中NMN含量都较少,如果想通过食物来补充NMN非常困难。所以近年来以NMN为主要活性物质的膳食补充剂成为消费市场的宠儿,但是诸多乱象也如影随形,其中最严重的就是部分NMN产品根本不含有NMN或者NMN含量远少于其标称,达到以次充好的目的。目前国内可以在跨境电商平台上购买到NMN跨境产品,但这类产品售价普遍十分高昂,最便宜的也要1500元/瓶,贵的甚至有20000元/瓶[14],如果没有做好产品质量控制,将严重损害消费者权益。所以2021年2月国家市场监管总局食品经营司下发《关于排查违法经营“不老药”的函》(以下简称“排查函”),明确指出目前NMN在我国未获得药品、保健食品、食品添加剂和新食品原料许可,不能作为食品进行生产和经营,要求各个省级市场监管部门对相关经营者进行全面排查。
目前国际上还没有NMN跨境产品中NMN含量测定方法标准,无法对此类产品中NMN含量进行准确测定,就无法对相关食品进行监管。所以非常有必要研制NMN跨境产品中NMN含量的检测方法标准,可以有效解决NMN行业中以次充好问题,实现净化市场的目的,并满足监管需求。我国是NMN原料的主要生产国之一,开发相关标准方法对于促进我国NMN的综合利用具有重要意义。
NMN具有极性大、不易挥发、分子内成盐、易溶于水、难溶于有机溶剂的性质[15−18],前人研究[19]认为,该性质制约了很多常规定量分析方法的应用。常见的定量检测方法包括液相色谱-紫外法(HPLCUV)[20]、液相色谱-质谱法(HPLC-MS)[21]、毛细管电泳法(CE)[22]、核磁共振波谱法(NMR)[23]。由于在NMN产品中NMN含量较高,通常都在10%以上,HPLC-MS、NMR方法灵敏度高、仪器昂贵,造成检测成本较高,而CE法装机量较少,应用还不够普遍。而HPLC-UV法以其操作简单、普适性好、应用广泛而成为NMN检测的利器。目前对NMN的定量检测主要集中于细胞提取物[24]、果蔬等基质[25],缺乏胶囊、片剂等剂型基质中NMN含量检测方法研究。有鉴于此,本文将通过优化样品的前处理方法和液相色谱方法,开发胶囊、片剂等剂型NMN跨境产品中NMN含量开发相应的检测方法,并通过方法学验证,为建立相应的检测方法标准奠定方法学基础。
1 材料与方法
1.1 材料与仪器
NMN标准品:β-烟酰胺单核苷酸(C11H15N2O8P)(CAS 1094-61-7,纯度≥98%) 上海源叶生物科技有限公司;甲醇(CH4O) 色谱纯,德国Merck公司;甲酸(CH2O2) 色谱纯,国药集团化学试剂有限公司;6种市售NMN胶囊、NMN片剂产品(其中4种为片剂、2种为胶囊剂) 采购于天猫。
iChrom5100分析型国产液相色谱仪,配DAD检测器 大连依利特分析仪器有限公司;Venusil HILIC色谱柱(4.6 mm×250 mm,5 μm,100 Å) 天津博纳艾杰尔科技有限公司;Thmorgan-VM200型涡旋振荡器 托摩根生物科技有限公司;HITACHICF15RXII离心机 日立公司;Sartorius分析天平 赛多利斯科学仪器(北京)有限公司;ELGA纯水仪 北京诚驿恒仪科技有限公司;LMDTC-15F超声波清洗仪 北京绿棉科技有限公司。
1.2 实验方法
1.2.1 溶液配制
1.2.1.1 标准储备液 准确称取10 mg NMN(精确到0.01 mg)标准品置于10 mL容量瓶中,用50%甲醇水溶解并定容至10 mL,配成浓度为1000 μg/mL标准储备液,避光保存于4 ℃冰箱备用,有效期3个月。
1.2.1.2 标准工作溶液 以50%甲醇水溶液为溶剂,将NMN标准储备液稀释至500、250、100、50、25、10、5 μg/mL的系列标准工作液,此溶液需现用现配。
1.2.2 样品前处理 取去除胶囊的粉末状样品100 g于石英研钵中,研磨至碎,过0.18 mm筛备用。
准确称取已研细的去除胶囊的粉末状样品0.5 g(精确到0.001 g)于50 mL容量瓶中,加入25 mL 50%甲醇水溶解,超声20 min使其充分溶解,提取两次,合并提取液,以10000 r/min的速度离心5 min,取上清液经0.22 μm有机滤膜过滤,取250 μL样品溶液用50%甲醇水定容至10 mL后进样检测。
片剂样品前处理方法同上。
1.2.3 液相色谱条件 液相色谱柱为Venusil HILIC(4.6 mm×250 mm,5 μm,100 Å),进样体积为10 μL,流速1 mL/min,柱温:35 ℃,检测波长为235 nm,流动相A为0.1%甲酸水溶液,流动相B为0.1%甲酸甲醇溶液,采用等度洗脱,流动相A:流动相B=15:85(V/V)。
1.2.4 标准曲线的绘制 将配制好的5、10、25 、50 、100、250、500 μg/mL标准工作溶液按“1.2.3”的液相色谱条件进样分析,以NMN的浓度为横坐标,峰面积响应值为纵坐标,绘制系列标准曲线,进行线性回归,得到NMN的标准曲线线性回归方程。
1.2.5 精密度试验 在相同仪器条件下,取90 μg/mL浓度的标准工作溶液,按“1.2.3”色谱条件重复进样6次,测得各被检测组分的峰面积,代入标准曲线线性回归方程得出测定浓度值,再计算测定浓度的相对标准偏差RSD。
1.2.6 稳定性试验 在相同仪器条件下,精密称取0.5 g(精确到0.001g)已粉碎的片剂样品以及胶囊样品以“1.2.2”的前处理方法,制成两种基质的提取液,在同一天内,以“1.2.3”色谱条件进样分析,每2 h测定一次,总共测定6次,测得被检测组分的峰面积,代入标准曲线线性回归方程得出测定浓度值,再计算测定浓度的相对标准偏差RSD。
1.2.7 重复性试验 在相同的仪器条件下,精密称取0.5 g(精确到0.001 g)已粉碎的片剂和胶囊样品6份,按“1.2.2”前处理方法方法操作,将处理好的待测液按“1.2.3”色谱条件进样分析,测得样品中NMN的峰面积,代入标准曲线方程,计算测定浓度的标准偏差RSD。
1.2.8 定量限、检出限的测定 将“1.2.1.2”的标准工作溶液按“1.2.3”的液相色谱条件测定,测得NMN的峰面积,代入相应物质的标准曲线回归方程中。以最低水平标准溶液中目标组分的3倍平均信噪比为检出限(LOD),10倍平均信噪比为定量限(LOQ),计算得本方法的LOD和LOQ。
1.2.9 加标回收率试验 准确称取0.5 g(精确到0.001 g)已粉碎的片剂样品、胶囊样品各12份,编号1~12,其中1~3号作为本底(基质样品),加入一定量标准储备液,4~6号为加标量为5 μg/mL的待测液,7~9号为加标量为10 μg/mL的待测液,10~12号为加标量为50 μg/mL的待测液,按照“1.2.2”操作,将处理好的待测液以及低、中、高3种浓度的加标样品经HPLC分析,测得峰面积,计算回收率。
1.3 数据处理
本文标准曲线线性回归方程及相关系数由依利特iChrom5100 workstation处理所得,其他数据采用Origin 6.0和Excel 2010软件对数据进行处理并作图,实验数据取3次平行实验的平均值。
2 结果与分析
2.1 液相色谱条件优化
经查阅文献[26−27],研究者们通常采用反相C18色谱柱来实现NMN的分离,但由于NMN的极性较大,几乎没有保留,出峰时间过早,容易与样品基质峰发生混淆。所以本文考虑使用对于极性样品分离效果较好的HILIC色谱柱[28−29]进行样品的分离。
为尽可能地将待测的NMN与胶囊和片剂样品基质中其他成分分离开,首先对流动相溶液配比进行了优化。用50%甲醇水溶液作为提取液溶解胶囊和片剂样品配制成待测液,分别以95% B、85% B、50% B(V/V)的等度分离方式对NMN胶囊和片剂样品进行色谱分离,通过单标法对峰的归属进行了确证,由此判断检测时最佳的流动相比例。分离谱图如图2所示,其中图2a为NMN片剂样品的分离谱图,图2b为NMN胶囊的样品分离谱图。从图中可以看出,随着流动相中甲醇含量的增大,NMN的出峰时间逐渐后移。对于片剂样品以50%比例等度洗脱时,NMN与基质无法得到有效分离,而使用95% B比例等度洗脱时NMN的出峰时间较晚,影响分析方法的效率,而且也伴随着峰展宽。当使用85% B比例等度洗脱时分离效果最好,NMN的出峰时间较短,峰形也基本满足高斯分布,可以达到基质中杂质峰与被分析物色谱峰分离的效果,降低了杂质峰干扰的可能,从而省去对提取液进行净化处理的步骤,可以将分析方法的总体时间控制在8 min内完成,使得操作简便、耗时较短,提高了分析检测效率。对于基质较为简单的胶囊样品,可以看出同样使用85% B比例等度洗脱,具有最好的分离效果、峰形和分析效率。所以采用85% B比例等度洗脱进行后续实验。

图2 NMN片剂样品基质(a)和NMN胶囊样品基质(b)的流动相优化分离色谱图Fig.2 Chromatograms of NMN tablet sample matrix (a) and NMN capsule sample matrix (b) based on mobile phase optimization
对于可离子化的样品,甲酸具有改善峰形的作用[30]。因此,本文对流动相中甲酸的加入进行优化对比,在85%比例等度洗脱条件下,考察了0.1%甲酸加入前后NMN片剂和NMN胶囊剂样品基质中各被测组分分离效果,结果如图3所示。结果表明,不加甲酸,NMN的峰拖尾现象明显,而且出峰时间明显推迟了8 min。而加入甲酸使各被测组分的峰形得到了良好的改善,并将整个分析时间控制在8 min内,极大地提高了分析效率。这可能是由于甲酸的加入抑制了NMN的解离,降低了其在HILIC色谱填料上的保留,缩短了出峰时间。所以在流动相中加入甲酸进行后续实验。
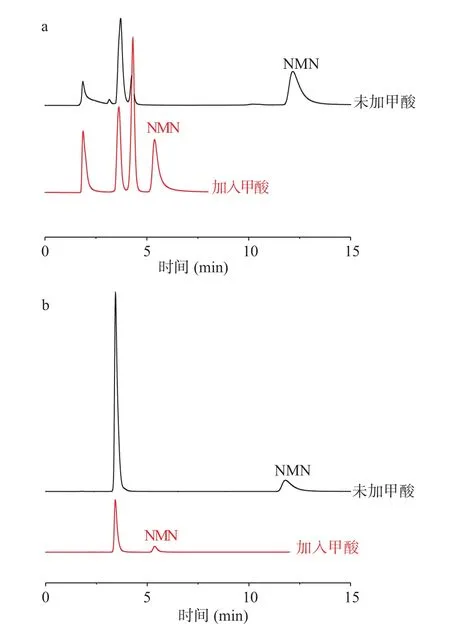
图 3 甲酸加入前后NMN片剂(a)NMN胶囊剂(b)样品基质分离谱图Fig.3 Chromatograms of NMN tablets matrix (a) and NMN capsule sample matrix (b) before and after formic acid addition
综上,对于NMN片剂和NMN胶囊基质,采用添加甲酸的方式能够改善NMN峰形,因此以0.1%甲酸水溶液-0.1%甲酸甲醇溶液(15:85,V/V)为流动相进行分离。
2.2 提取溶剂优化
根据NMN的理化性质[10−12],NMN在水中溶解度较大,在甲醇中溶解度较小,在仪器及实验条件相同情况下,本实验选择了水溶液、50%甲醇水溶液、85%甲醇水溶液3种溶剂开展样品提取溶剂优化实验。由于NMN在甲醇中几乎不溶,故不选择用纯甲醇作为提取溶剂。取0.5 g粉碎后的片剂和胶囊样品,向其中各添加一定量的NMN标准储备液,使加标浓度为10 μg/mL,采用“1.2.2”的样品提取方式,分别以上述3种溶剂进行提取,并测定加标回收率,结果如下表1所示。从表1中可以看出,以50%甲醇水溶液作为提取溶剂,样品的回收率可控制在90%~110%之间,同时偏差最小,具有最佳的提取效果。因此,在后续实验中采用50%甲醇水提取溶剂进行提取。
2.3 标准曲线、检出限、定量限
以“1.2.3”中的色谱分析条件对NMN标准工作溶液进行测定,通过iChrom5100 workstation绘制标准曲线回归方程,其线性范围、相关系数、LOD及LOQ如表2所示。
由表2数据可知,本方法在5~500 μg/mL范围内有较好的线性关系,相关系数r为0.999。经计算,NMN的检出限(LOD,S/N=3)为1.0 mg/kg,定量限(LOQ,S/N=10)为3.0 mg/kg。
2.4 仪器精密度实验
按照“1.2.5”中实验步骤,对浓度为90 μg/mL的NMN标准溶液进行6次平行测定,将测得的NMN的响应值带入线性方程计算得到溶液中NMN的含量,并计算6次测定结果的RSD如表3所示。
由表3数据可知,仪器精密度实验结果的RSD为0.3%,表明本文发展的液相色谱方法具有较好的重现性,方法精密度较好,可以用于后续实际样品中N MN含量的测定。
2.5 稳定性实验
以“1.2.6”中实验步骤进行操作,考察方法的稳定性,经过6次平行实验,测得片剂和胶囊样品中NMN的浓度与RSD如表4所示。
由表4数据可知,对于NMN片剂和NMN胶囊,6次测定结果RSD均较小,说明结果都保持高度一致,可以保证方法的稳定性。
2.6 重复性实验
按照“1.2.7”中的实验步骤,将测得的NMN片剂、胶囊原液的6组平行样品中NMN的浓度及RSD计算汇总,结果如下表所示。
由表5数据可知,两种样品重复性实验的RSD在0.6%~1.8%之间,表明本方法重复性较好,表明本文开发的前处理方法结合HPLC方法可以用于NMN片剂和NMN胶囊中的NMN含量测定,并能使结果具有良好的重复性。
2.7 加标回收率实验
按“1.2.9”中步骤进行操作,对制备的5、10、50 mg/kg的低、中、高三种加标水平的NMN片剂和NMN胶囊剂样品中NMN含量进行了测定,并计算回收率和RSD%,结果如表6所示。
由表6数据可知,NMN片剂和NMN胶囊样品中NMN的检测回收率在90.9%~108.9%之间,相对标准偏差均不超过1.9%,表明本方法回收率好,结果准确可靠,本方法可用于实际样品中NMN含量的准确测定。
2.8 实际样品测定
按照本文发展的NMN片剂和NMN胶囊剂样品基质中NMN含量的检测方法对市售6个品牌的NMN产品中的NMN含量进行分析测试,检测结果见表7。
实际样品分析结果表明,所有样品中都检出了NMN,但是部分产品的实际含量较其标称含量偏少,如2号样品中NMN含量只有其标称值的一半,3号样品中NMN含量只有其标称值的三分之一,涉及虚假宣称。

表 1 不同溶剂对NMN提取效率的影响(n=3)Table 1 Effects of different solvents on the extraction efficiency of NMN (n=3)

表 2 NMN的线性范围、LOD和LOQTable 2 Standard curve, linear range, LOD and LOQ of NMN

表 3 精密度实验结果(μg/mL)Table 3 Precision test results (μg/mL)

表 4 稳定性实验结果(mg/kg)Table 4 Results of stability experiments (mg/kg)

表 5 重复性实验结果(mg/kg)Table 5 Results of repeatability experiment (mg/kg)
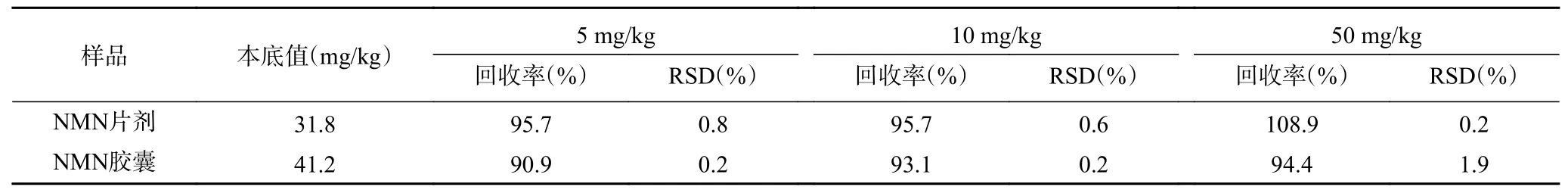
表 6 加标回收率实验结果Table 6 Experimental results of standard recovery

表 7 6种不同NMN产品中NMN含量(g/kg,n=3)Table 7 NMN content in 6 different NMN products(g/kg, n=3)
3 结论
本文建立了一种NMN胶囊和NMN片剂基质中NMN含量的检测方法,方法通过调节色谱分离条件,使目标组分与基质成分完全分离,无需进行样品净化操作,同时控制流动相组成,整个分析时间控制在8 min内,具有操作简便、检测效率高、准确性好等优点。该方法为制定NMN跨境产品中NMN含量的检测方法标准奠定了方法学基础,对于提高NMN相关产品质量,促进相关产业的便捷监管具有深远意义。
猜你喜欢
核化学与放射化学(2022年2期)2022-04-28
中国土壤与肥料(2021年5期)2021-12-12
中国土壤与肥料(2021年5期)2021-12-02
饲料博览(2020年7期)2020-08-18
北京园林(2020年4期)2020-01-18
商品与质量(2019年15期)2019-11-28
现代职业教育·高职高专(2019年7期)2019-09-20
现代园艺(2019年3期)2019-03-08
医学研究杂志(2015年12期)2015-06-10
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
