
谷歌带来蛋白质领域新突破,一举注释近百分之十蛋白质序列
2022-04-29 17:58
海外星云 2022年7期
近日,谷歌训练出一种名为ProtCNN的深度学习模型,其可以用于准确预测蛋白质序列的功能,使更多未知蛋白质序列得到注释。据了解,这些注释是基于主流蛋白质家族数据库Pfam构建的严格基准所进行的评估,Pfam数据库记录了一系列蛋白质家族及其功能注释。
该研究的成功,令Pfam数据库中蛋白质序列的覆盖范围扩大了9.5%,超越了过去十年里科学家在此方面的成果,并预测了360种Pfam数据库未注释过的人类蛋白质的功能。
相关论文以《使用深度学习来注释蛋白质宇宙》为题发表在Nature Biotechnology上。

相关论文
伴随DNA测序成本的降低和宏基因组测序项目的兴起,具有蛋白质序列功能注释作用的高效工具对生物技术的发展愈加重要。
此前,常用的蛋白质序列功能的注释方法是,在大型标记序列集合上进行成对比对的BLASTp查詢方法和基于signature构建的profile隐马尔可夫模型。
这些方法虽然是有用的,效率却相对较低。过去5年里,使用这些方法仅让Pfam数据库中蛋白质序列数量增长了不到5%。
为此,谷歌的研究人员对深度学习模型是否可以补充现有方法进行探究,并提供了能够更广泛覆盖蛋白质宇宙的蛋白质序列功能预测模型ProtCNN。
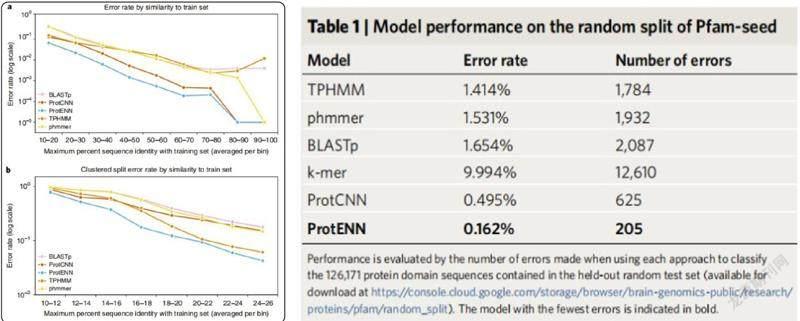
ProtCNN模型性能的表现
在蛋白质序列的比对过程中,测试数据和训练数据之间会存在相似性,这意味着模型性能必须根据每个保留的测试序列与训练序列之间的相似性作分层。
通过分析蛋白质序列中的随机和聚类分裂,这里的序列是使用基于相似性的聚类成员而分配给测试或训练分裂的,研究人员发现,在注释保留的测试序列时,ProtCNN模型比当前方法在随机和聚类分裂中产生的错误更少。
为确认模型捕捉到了未对齐蛋白质序列的结构,研究人员使用跨蛋白质家族学习的联合表示法,一次性学习注释模型未训练蛋白质家族的序列。
此外,要为更多蛋白质序列集带来注释,还须进行远程同源性检测。远程同源性检测是指准确分类训练数据集中并不相似的蛋白质序列。
而将ProtCNN模型与现有方法相结合,则大大提高了远程同源性检测的准确性,这对扩大蛋白质宇宙的覆盖范围至关重要。
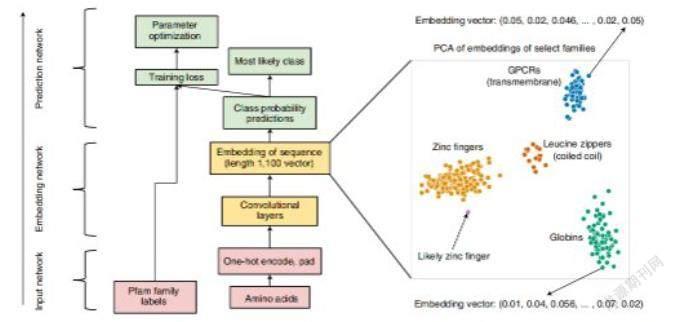
ProtCNN模型的架构
研究结果证实,今后,类似ProtCNN的深度学习模型将是蛋白质序列功能注释研究中的核心支撑技术。
作为一个以蛋白质序列为研究对象的计算生物学者,来自美国德克萨斯大学西南医学中心的助理教授丛倩评价该研究道,“我很高兴看到优秀的生物科学和计算机科学工作者在这个领域做出的尝试和贡献。”
她表示,该研究主要探讨了通过序列预测蛋白质功能,相比于当下正在创造历史的三维结构预测,这一问题对人工智能来说更有挑战性,并总结了具体原因。
对于蛋白质功能预测困难的原因,丛倩进行了如下总结。
首先,蛋白质功能的可靠数据量并不大,且功能不像结构那样容易被量化。其次,一般来说相似序列的蛋白质在三维结构上也区别不大,但其功能却很有可能大相径庭。最后也是最重要的一点,即严格来讲,在AlphaFold问世之前,已经没有严格意义上的全新三维结构了。所谓的“新”蛋白质结构都是已知结构的简单组合,如果人工智能算法掌握了所有已知结构,其将有能力推导出任何一个“新”蛋白质序列的结构。
另外,丛倩补充说,自然界中的蛋白质应该还有很多未发现的新功能,而人工智能算法很难在现有技术框架下预知这类从未见过的功能。

丛倩
不过在谷歌的这项新研究中,其并非在解决预测新功能的问题,而是想要更广泛、更精确地发现同样具有某种已知功能的其他蛋白质,这有助于人类快速了解一个新物种中绝大多数蛋白质的作用,如跟人类疾病相关或是具有潜在工业价值的蛋白质。
丛倩称,她对蛋白质领域的这些研究相当感兴趣,但对于人工智能是否在这个领域比传统方法更可靠的问题,其仍存有疑虑。
她表示,“我曾经有几个梦想。第一,通过序列准确的预测蛋白质的结构;第二,通过序列准确预测蛋白质之间的相互作用;第三,通过序列准确预测蛋白质的功能。”得益于人工智能的飞速发展,其梦想或是已经实现,或是在不久的将来即将实现。
目前,丛倩所在实验室的主要目标正是通过解决第二个问题来辅助解决第三个问题。她说,事实上,第三个问题才是真能带来全新科学发现,也是像她这样的科学工作者最感兴趣的问题,其将会在其项目上尝试这方面的研究。
丛倩表示,希望未来有更多科学家把注意力放到类似更有挑战性的问题上,带领人工智能去探索更难定义、更难量化的领域是我们这代人的历史使命。(综合整理报道)(编辑/多洛米)
猜你喜欢
今日农业(2022年15期)2022-09-20
中学生数理化·高二版(2022年4期)2022-05-09
中学生数理化·高二版(2022年4期)2022-05-09
爱你·健康读本(2020年4期)2020-04-30
财经(2017年2期)2017-03-10
财经(2016年15期)2016-06-03
财经(2016年3期)2016-03-07
财经(2016年6期)2016-02-24
金点子生意(2014年4期)2014-04-10
中学生英语高效课堂探究(2008年9期)2008-11-17
