
Planar Tetracoordinate Carbon in 6σ + 2π Double Aromatic CBe42- Derivatives①
2022-04-16 02:59JINBoBIANJinHongZHAOXueFengYUANCiXiGUOJinChngWUYnBo
结构化学 2022年3期
JIN Bo BIAN Jin-Hong ZHAO Xue-Feng YUAN Ci-Xi GUO Jin-Chng WU Yn-Bo②
a (Key Laboratory of Materials for Energy Conversion and Storage of Shanxi Province,Institute of Molecular Science, Shanxi University, Taiyuan 030006, China)
b (Department of Chemistry, Changzhi University, Changzhi 046011, China)
ABSTRACT As a typical electron deficient element, beryllium is potentially suitable for designing the species with novel non-classical planar hypercoordinate carbon due to high preference for the planar structures by small beryllium-containing clusters. In particular, the CBe54- cluster with a planar pentacoordinate carbon (ppC) had been proved by many previous studies to be an excellent template structure for the systematic design of ppC species through attaching various monovalent atoms on the bridging position of Be-Be edges. In this work, based on the analysis and extension on our recently reported CBe4Mnn-2 (M = Li, Au, n = 1~3) species, we propose that ptC cluster CBe42- is similar to CBe54- in that it can also be employed as a template structure to systematically design the ptC species through binding various monovalent atoms on the bridging position of Be-Be edges. Our extensive screening suggests that the feasible bridging atoms (E) can be found in group 1 (H, Li, Na), group 11 (Cu, Ag, Au),and group 17 (F, Cl, Br, I) elements, leading to total thirty eligible ptC species with CBe4 core moiety (CBe4Enn-2).The ptC atoms in these species are involved into three delocalized σ bonds and a delocalized π bond, thereby not only obeying the octet rule, but also possessing novel 6σ +2π double aromaticity, which significantly stabilizes the ptC arrangement. In addition, the attached bridging atoms can stabilize the CBe4 core ptC moiety by replacing the highly diffused Be-Be two-center two-electron bonds with the much less diffused Be-E two-center two-electron bonds or Be-E-Be three-center two-electron bonds, as reflected by the increasing HOMO-LUMO gaps when the number of bridging atoms increases. Remarkably, the stochastic search algorithm in combination with high level CCSD(T) calculations revealed that twenty-six of the thirty-one ptC species (including previously reported six species) were global energy minima on their corresponding potential energy surfaces, in which twenty-five of them were also confirmed to be dynamically viable. They are suitable for the generation and characterization in gas phase experiments and followed spectroscopic studies.
Keywords: planar hypercoordinate carbon, aromaticity, beryllium, DFT calculations, global energy minimum; DOI: 10.14102/j.cnki.0254-5861.2011-3332
1 INTRODUCTION
As a typical electron deficient element (two electrons in four valence orbitals), beryllium (Be) prefers the multicenter bonding to have the number of its valence shell electrons as much as possible. Such a bonding preference is particularly useful for designing the species with non-classical planar hypercoordinate carbon (phC). In 2008, Wang group first designed a series of planar tetracoordinate carbon(ptC) species involving beryllium[1]. In 2010 and 2016, Wu and Wang groups designed BeH-based ptC species with the carbon skeletons of monocyclic arenes and ethylene, whose structural models were also extended to various types of nanomolecules[2,3]. In 2018 and 2019, Guo et al. reported a series of global minima ptC species, including CBe4Au4,CBe4Li4, and CBe3X3+(X = H, Li, Na, Cu, Ag)[4-6].
In comparison with the being employed for designing ptC species, beryllium was more frequently and successfully employed in designing species with planar pentacoordinate carbon (ppC) previously. In 2010 and 2012, Wang, Wu, and Merino joint groups reported the global energy minima(GEM) ppC species CAl4Be, CAl3Be2-, CAl2Be32-, and CAl2Be3Li-by substituting the aluminum atom(s) in milestone CAl5+with beryllium atom(s)[7,8]. In 2018, Wu group further found that these GEM ppC species can be further stabilized by introducing H atom on the peripheral edges involving beryllium atom[9]. In 2012, Merino group designed the GEM CBe5E-(E = Al, Ga, In, Tl) with a CBe4E ppC core moiety[10]. In 2008, Luo et al. reported the hexatomic species CBe5and CBe54-with a ppC[11]. Remarkably, though highly negatively charged CBe54-was unstable, it was employed subsequently as a core moiety to design dozens of σand π double aromatic GEM species with a ppC CBe5Enn-4(n=1~5) by introducing various kinds and numbers of monovalent elements (including H, Li, Na, K, F, Cl, Br, Au) to the bridging positions of Be-Be edges[12-15].
Beryllium has also been applied to design the species with planar hexacoordinate carbon or boron (p6C[16]or p6B[17]) as well as the planar heptacoordinate transition metals. Beryllium was utilized to design the monolayer sheets with perfect ptC[18], the zig-zag double chain nanoribbon with ppC and ribbon aromaticity[19], the quasi-ppC-containing monolayer sheet featuring the half metallic property and negative Poisson ratio[20], and the semi-conductive monolayer sheet with quasi-p6C[21].
In the application of beryllium to design phC species, the most exciting part by far should be the design of ppC, because beryllium was involved in more than half of GEM ppC species. In particular, the ppC-containing CBe54-derivatives were the paragon of such non-classical molecule design. Dozens of species can be designed by stepwise introduction of various mon-valent elements onto the Be-Be edges of CBe54-and the majority of these species were turned out to the GEM. Is there a similar paragon that can be employed in the design of ptC?The answer is positive. We will show in the following that CBe42-species can play the parallel role in designing ptC species to that played by CBe54-in designing ppC species.
2 COMPUTATIONAL METHODS
The geometries of designed species were optimized and the subsequent harmonic vibrational frequencies were analyzed at both B3LYP/BS1 and B2PLYP-D/BS1 levels. Herein, BS1 is a mixed basis set used in this work with aug-ccpVTZ for elements lighter than Cu and aug-cc-pVTZ-PP for Cu and heavier elements; while B2PLYP-D is a double hybrid B2PLYP functional considering the empirical dispersion. The geometries and frequencies obtained using B3LYP functional were not different in essential from those obtained using B2PLYP-D, so the B3LYP functional was employed for further studies, including electronic structure analyses and stability explorations. The chemical bonding in the designed species was interpreted using adaptive natural density partitioning (AdNDP) analyses at the B3LYP/BS2 level,where BS2 denotes a mixed basis sets with 6-31G(d) for elements lighter than Br and LANL2DZ for heavier elements.The potential energy surfaces of the concerned components were explored using the stochastic search algorithm. Both the singlet and triplet surfaces were considered. The generated random structures were initially optimized at the B3LYP/BS2 level, and then ten lowest isomers were optimized and harmonic vibrational frequencies were analyzed at the B3LYP/BS1 level. The energies of these isomer were further improved at the CCSD(T)/BS1 level and the relative energies were compared using the CCSD(T)/BS1 single point energy in combination with the zero-point energy corrections obtained at the B3LYP/BS1 level (abbreviated as CCSD(T)//B3LYP). The dynamic stability of designed species was studied using Born-Oppenheimer molecular dynamic (BOMD)[22,23]simulations at the B3LYP/BS2 level at 298 K. The structural evolution during simulation was described by the root-mean-square derivation (RMSD, in Å)relative to the B3LYP/BS2-optimized structures. The nucleusindependent chemical shifts (NICS)[24-26]were calculated at the centers of the three-membered rings and the points located 2 Å above the centers of the three-membered rings and above ptCs to assess the aromatic nature of these ptC species. The AdNDP analyses were performed using AdNDP program[27,28], the stochastic search algorithm[29,30]was realized using GXYZ 2.0 program[31,32], the CCSD(T) calculations were carried out using MolPro 2012.1 package[33], and all other calculations were performed using Gaussian 16 package[34].
3 RESULTS AND DISCUSSION
We had recently reported the ptC species CBe4Mnn-2(M =Li, Au,n= 1~3)[35]. In that work, we focused on achieving the 14 valence electrons species with ptC rather than the systematic design of ptC species based on a good template structure. Nevertheless, we found that the similar structural model might be extended to the congeners of Li and Au, including Na, Cu, and Ag, though the potential energy surfaces of corresponding species were not explored. Therefore, consulting the previous excellent works on designing ppC species based on CBe5core moiety, we speculated that CBe4might be another excellent core moiety for systematically designing new ptC species by attaching the monovalent atoms on the Be-Be edges. First, we verify the molecular charges of CBe4cluster,which can be deduced from our previously reported species having the formula of CBe4Mnn-2(n= 1~3). If the value ofnis extended to zero, a dianionic cluster with the formula of CBe42-(0, see Fig. 1) can be obtained. At both B3LYP/BS1 and B2PLYP-D/BS1 levels, such a cluster adopts the planar structure inC2vsymmetry and encloses a ptC. As a comparison,neutral CBe4and the highly negatively charged CBe44-adopt the non-planar structures inD2andD2dsymmetries, respectively, and thus they are disregarded in the following. The C-Be interatomic distances in 0 are 1.60 and 1.68 Å, short enough for each beryllium atom to be counted as a coordination to center carbon, so 0 is an eligible ptC species.
The AdNDP analysis revealed that 0 had seven pairs of valence electrons, in which three of them were highly diffused Be-Be two-center two electron (2c-2e) σ bonds with occupation number of 1.97 |e|. The diffusion character of such σ bonds, possibly attributed to the high negative charges of the molecule and the metallic nature of beryllium, is unbeneficial to the stabilization of 0 because the electrons at the position far from the nuclei will be reactive. Therefore, the elimination of such diffused σ bonds is highly desired. In contrast, though the remained four pairs of electrons involve all five atoms, they are less diffused than the Be-Be 2c-2e bonds. Note that such four 5c-2e bonds (ON = 2.00 |e|) supported eight electrons to valence shell of the central carbon,which stabilizes the molecule by making the carbon meet the octet rule. In addition, four pairs of electrons consist of three 5c-2e σ and one 5c-2e π bonds, matching the famous 4n+2 rule withn= 1 and 0, respectively, so the preference of ptC structure in 0 may stem from the novel σ and π double aromaticity. Therefore, retaining such 5c-2e bonds is also highly desired. Remarkably, according to our extensive exploration of CBe42-potential energy surface, 0 is GEM and locates 4.17 kcal·mol-1lower in energy than the second lowest isomer (0a, see Fig. S4) at the CCSD(T)//B3LYP level. As a comparison, CBe54-ppC structure is a high energy local minimum. The lower molecular charge and the GEM nature of 0 suggest its higher plausibility as a core moiety than CBe54-species for the subsequent design of phC species.

Fig. 1. Optimized structure of CBe44-, CBe42- (0), and CBe4 at the B3LYP/aug-cc-pVTZ level (A) and the AdNDP view of chemical bonding in 0 (B)
The systematic design of the ptC species was realized by attaching the monovalent element on the bridging positions of 0, similar to the design of ppC species based on CBe54-.The literature survey had revealed the feasible bridging atoms (E) that had been employed for the ppC design based on CBe54-, including group 1 elements H, Li, Na, group 11 elements Cu, Ag, Au, and group 17 elements F, Cl, Br, I. In this work, the design of ptC species involves the stepwise introduction of these monovalent E elements onto the Be-Be edge(s). As shown in Scheme 1, for the species with one or two E atoms,i.e. CBe4E-and CBe4E2, we considered two possible structural models, which are labelled as M1/M1′and M2/M2′, respectively. Note that a new model can be found for both CBe4E-and CBe4E2species (M1″ and M2″,see Scheme 1) during the isomer search.
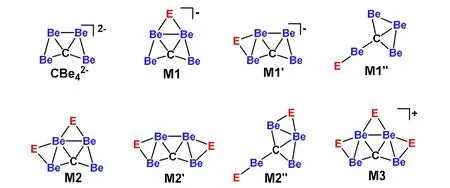
Scheme 1. Stepwise introduction of E atom(s) on the Be-Be of CBe42-
We have computed the isomers in various model structures. Fig. 2 shows those with lower energies. If E is an alkali or group 11 metal, it tends to bind CBe4moiety on the top Be-Be edge (M1 model) of CBe42-structure (see CBe4E-(E= Li (1), Na (4), Cu (7), Ag (10), and Au (13)) in the first column of Fig. 2) rather than on the side Be-Be edge (M1′model). If E is H or a halogen, it prefers neither top nor the side Be-Be edges, but prefers the terminal position on a bottom Be atom (M1″ model) (see CBe4E-(E = H (16), F (19),Cl (22), Br (25), and I (28)) in the fourth column of Fig. 2).Note that the ground electronic state for 19 is triplet (E = F)and that for other CBe4E-species is closed-shell singlet.
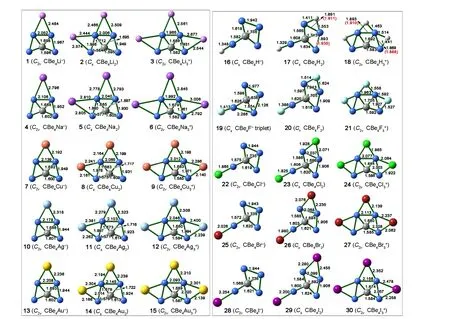
Fig. 2. Optimized structures of ptC species designed based on 0. The necessary bond lengths regarding molecular symmetries are given in Å
When two E atoms are anchored on the CBe4moiety to give CBe4E2species, the situation is similar in structural preference to CBe4E-: If E is an alkali or group 11 metal,two E atoms tend to bind CBe4moiety with one E atom on top and the other on the side Be-Be edges (M2 model), respectively (see CBe4E2(E = Li (2), Na (5), Cu (8), Ag (11),and Au (13)) in the second column of Fig. 2) rather than both on the side Be-Be edges (M2′ model). As a comparison, if E is H or a halogen, corresponding CBe4E2species prefer the M2″ model (see CBe4E2(E = H (17), F (20), Cl (23), Br (26),and I (29) in the fifth column of Fig. 2) with one E atom on side Be-Be edge and the other E atom on the terminal position of a bottom Be atom. The ground electronic state of all CBe4E2-species is closed-shell singlet.
Nevertheless, when three E atoms bind the CBe4moiety,corresponding CBe4E3+species always prefer the M3 model structure (Scheme 1) despite the type of E atom. The third and sixth columns of Fig. 2 show the structures of these 10 species,i.e.CBe4E3+(E = Li (3), Na (6), Cu (9), Ag (12), Au(15), H (18), F (21), Cl (24), Br (27), and I (30)).
As Fig. 2 shows, all these thirty species adopt planar structures with carbon atom surrounded by four Be atoms.The C-Be distances in 1~30 range from 1.568 to 1.736 Å,shorter than the sum of covalent radii of C and Be (1.790 Å),so the carbon atoms in 1~30 are the well-defined ptCs. The Be-Be distances in 1~30 range from 1.869 to 2.208 Å,covering the typical lengths of Be-Be single[36], double, and pseudo-triple bonds[37,38]. Remarkably, there are three very short Be-Be distances in CBe4H2(2) and CBe4H3+(3)(1.893/1.893, 1.891 and 1.869 Å, respectively), within the range of ultrashort metal-metal distances (USMMDs, defined asdM-M< 1.900 Å)[39]and suggesting the strong Be-Be interactions. We further re-optimized the geometries of CBe4H2(2) and CBe4H3+(3) to verify the USMMDs at the CCSD(T) level and corresponding geometric parameters are given in Fig. 2 in the parenthesis and red font. As Fig. 2 shows, the above ultrashort Be-Be distances were elongated slightly to 1.935/1.910, 1.911, and 1.888 Å, respectively after the optimization at the CCSD(T) level and only a Be-Be distance (1.888 Å) in 3 was still shorter than 1.900 Å and can be regarded as the USMMD. The USMMD of 1.888 Å can be rationalized by electronic analysis.

Fig. 3. AdNDP view of chemical bonding in CBe4Cu- (7), CBe4Cu2 (8), and CBe4Cu3+ (9) as well as CBe4H- (16), CBe4H2 (17), and CBe4H3+ (18)
The chemical bonding in these ptC species can be described by the AdNDP analysis. The AdNDP results for CBe4Cunn-2and CBe4Hnn-2(n= 1~3) are shown representatively in Fig. 3, while those for other species are similar to E= Cu or E = H, thus they are given in the Supporting Information. As Fig. 3 shows, in CBe4Cunn-2(n= 1~3) species, a diffused Be-Be 2c-2e σ bond with ON of 1.93 or 1.87 |e| can be found for the Be-Be edges without bridging Cu atom (see the first column in upper part), while a less diffused Be-Cu-Be 3c-2e σ bond with ON of 1.84~1.98 |e| can be found for the Be-Be edges with a Cu atom (see the second column in upper part). As a comparison, the peripheral bonding in CBe4Hnn-2(n= 1~3) is a little different in that there is a Be-H 2c-2e σ bond with ON of 2.00 |e| covering the terminal Be-H group of CBe4H-and CBe4H2. Other peripheral bonding patterns are similar to that in CBe4Cunn-2(n= 1~3) species, including a diffused Be-Be 2c-2e σ bond (ON = 1.84 or 1.93 |e|) on Be-Be edges without H atom and a less diffused Be-H-Be 3c-2e σ bond (ON = 1.98 or 1.99 |e|) on Be-Be edges with an H atom. Remarkably, the bonding patterns concerning the core CBe4moieties in CBe4Cunn-2and CBe4Hnn-2(n= 1~3) species are similar as they possess three CBe45c-2e σ bonds (ON = 1.97~1.99 |e|) and one 5c-2e π bond (ON = 1.97~1.99 |e|). These four orbitals are also similar to that in CBe42-template molecule and provide the basis for adopting the ptC structures. By meeting our initial expectation on the electronic structures of derived species (stepwise eliminating the diffused Be-Be σ bonds but retaining four central 5c-2e bond), the designed species would possess the better and better stability when number of E atoms(n) increases. In addition, the AdNDP results can rationalize the existence of USMMD in 18. As shown in Fig. 3, there are five orbitals contributed positively to shorten the concerned Be-Be distance, including one Be-H-Be 3c-2e σ bond, three CBe45c-2e σ bonds and one CBe45c-2e π bond. Please note that this does not mean that there is a pseudo-quintuple bond between two beryllium atoms because each of the four 5c-2e bonds only contributes partially for such Be-Be bonding.

Fig. 4. NICS values of CBe4Cu3+ (A) and CBe4H3+ (B). Along the vertical line, the NICS points (red balls) are 0.5 Å apart from their neighbors
As mentioned above, both the CBe42-template molecule and its derivatives retain three 5c-2e σ bonds and one 5c-2e π bond.Such an electronic structure not only leads to the planarity, but also corresponds to six delocalized σ electrons and two delocalized π electrons, meeting the Huckel’s 4n+ 2 rule forn= 1 and 0, respectively and suggesting the novel 6σ + 2π double aromaticity. To corroborate the existence of aromaticity, the NICS calculations were performed by using the highly stable CBe4E3species as the examples. The results for CBe4Cu3+(9)and CBe4H3+(18) are shown representatively in Fig. 4, while those for other species are given in Supporting Information. As shown in Fig. 4, the centers of Be-C-Be triangles in 9 possessed the NICS values of -25.5 and -18.8 ppm, while those in 18 showed the values of -24.7 and -21.6 ppm, suggesting the existence of σ aromaticity in both 9 and 18. Simultaneously,the points located 1.0 Å above the molecular plane all possessed the negative NICS values, ranging from -10.1 to -22.4 ppm and suggesting the existence of π aromaticity.
The stabilization effect of bridging E atoms can be verified by the variation of HOMO-LUMO gaps (Gap). The value for CBe42-is only 0.53 eV at the B3LYP/BS1 level. As shown in Fig. 5, the CBe4E-and CBe4E2species possess the obviously enlarged Gaps of 1.37~1.88 eV and 1.30~2.42 eV, respectively. Moreover, when the diffused Be-Be 2c-2e bonds are completely eliminated in CBe4E3+species, the Gaps increased dramatically to 2.38 to 4.59 eV. Except that E is an alkali metal (Li and Na), other CBe4E3+species possess the Gaps higher than 3.47 eV. Especially for E = H and halogens,the Gaps are all wider than 4.21 eV, revealing their welldefined electronic structures. The difference between H/halogens and alkali/group 11 metals mainly lies in their electronegativity. From electronic structure point of view, H can be considered as a halogen because it needs only one electron to have its valence shell occupied fully. When the Be-Be edges of CBe4core were surrounded by H and halogens (the typical non-metals), the resulted species should possess better electronic structures than the species whose Be-Be edges of CBe4core were surrounded by the alkali and group 11 metals. Consistently, the HOMO-LUMO gaps of the former are wider than those of the latter.
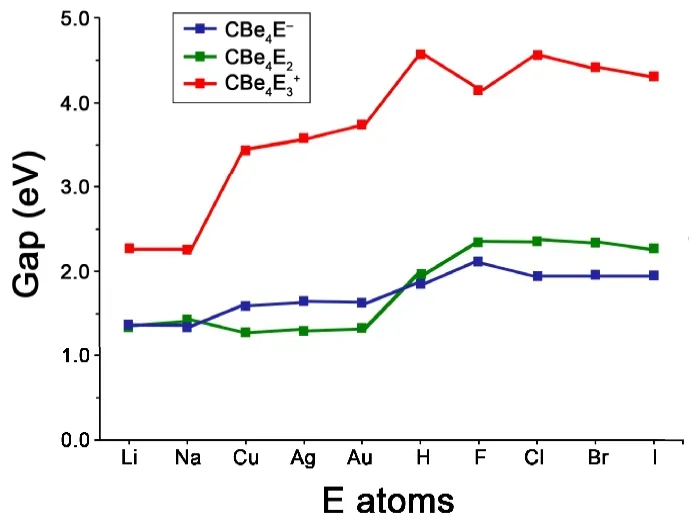
Fig. 5. Variation of HOMO-LUMO gaps (Gap, in eV) with the increasing number of E atom
To assess the possibility to realize the ptC species designed in this work, the systematic exploration of potential energy surfaces (PESs) was carried out using the stochastic search algorithm. According to the relative energy determination at the final CCSD(T)//B3LYP level, only the GEMs of CBe4E2(E = H, F, Cl, Br, I) components did not adopt the ptC structure, while those of all other CBe4Enn-2(E = H, Li,Na, Cu, Ag, Au, F, Cl, Br, I;n= 1~3) components adopted one of the ptC models shown in Scheme 1. As shown in Fig.S4, S5, and S6 in the Supporting Information, these GEMs located 0.6 to 21.8 kcal·mol-1below their second lowest isomers at the CCSD(T)//B3LYP level, suggesting their good thermodynamic stability.
The dynamic stability is equally important for the experimental viability of computationally designed species. In this work, the dynamic stability was studied using 100 picoseconds BOMD simulations for 26 GEM ptC species (including 0) at the B3LYP/BS2 level. The results for CBe42-(0), CBe4Cu3+(9), CBe4F3+(21), and CBe4H3+(18) are shown representatively in Fig. 6, while those for other GEMs are given in Figs. S7, S8, and S9 in the Supporting Information. We found that the RMSD plots for many of GEM ptC species showed upward jumps, suggesting the significant changes in the position of atoms. The detailed structural examination revealed that the geometry of CBe42-shows the reversible isomerization to a structure (0c) we located during the PES exploration. As Fig. 6 shows, 0c took up for the larger proportion during the simulation than 0, but the reversible transformation still suggested the high viability of 0 and indicated the high flexibility of Be atoms in 0.
The RMSD plot for the simulation of CBe4Ag2(11) shows the irreversible upward jump, which corresponds to the structural evolution to an isomer having a tetrahedral carbon(see 11a in Fig. S5), so it is dynamically unviable. That of CBe4Cu3+(9) shows some reversible and fleeting upward jumps. However, the detailed structural sampling revealed that the upward jumps corresponded to the flexibility of the molecule as a whole rather than the isomerization. The RMSD plot for CBe4F3+(21) does not show the upward jump and the fluctuation of RMSD values is small, suggesting the rather rigid structure of 21. Interestingly, the RMSD plot for CBe4H3+(18) shows two stepwise upward jumps, which correspond to two times of isomerization. In the first isomerization, the structure of 18 is converted to its second lowest isomer (see 18a in Fig. S6), while in the second isomerization, 18a is converted to 18 again. As shown in Fig. 6, if the labels (numbering of the atoms) are considered, we can see a Be-H group rotate around the central ptC during two times of isomerization. In the majority of simulation time, the structure of 18 is adopted, so 18 is also dynamically viable. The plots for CBe4Li2(2), CBe4Na2(3), CBe4Cu2(8), and CBe4I3+(30) are similar to that of 18, thus though they are not dynamically stable, they are still dynamically viable. The RMSD plots of other ptC GEMs are similar to 9 or 21, thus they are dynamically stable and viable.

Fig. 6. RMSD versus simulation time for the BOMD simulation of CBe42-, CBe4Cu3+, CBe4F3+, and CBe4H3+, respectively at the B3LYP/BS2 level and 298 K
4 CONCLUSION
In summary, we had proved that the strategy for designing ppC species based on CBe54-can be transferred to design the ptC species based on CBe42-. The monovalent atoms including group 1 elements H, Li, Na, group 11 elements Cu, Ag,Au, and group 17 elements F, Cl, Br, I are all suitable for further stabilizing CBe42-by anchoring the monovalent atoms on its Be-Be edges to stepwise eliminate the highly diffused Be-Be 2c-2e bonds. In thirty-one designed ptC species (including CBe42-itself), twenty-five of them are dynamically viable global energy minima, which are potentially possible for experimental realization in experiments. Remarkably, the dynamic simulations suggest that the Be and E atoms in some ptC species are highly flexible but the basic ptC geometry can be maintained during the changes of atom positions, thus suggesting the stable but flexible bonding nature.

- 结构化学的其它文章
- Structural and Electronic Properties of Lutetium Doped Germanium Clusters LuGen(+/0/-) (n = 6~19):A Density Functional Theory Investigation①
- Discovery of Benzimidazole Derivatives as Novel Aldosterone Synthase Inhibitors: QSAR, Docking Studies, and Molecular Dynamics Simulation①
- QSAR Models for Predicting Additive and Synergistic Toxicities of Binary Pesticide Mixtures on Scenedesmus Obliquus①
- Preparation, Crystal Structure and Fungicidal Activity of N-(5-(benzofuranol-7-oxymethyl)-1,3,4-thiadiazol-2-yl)amide Compounds①
- Antibiotic Silver Particles Coated Graphene Oxide/polyurethane Nanocomposites Foams and Its Mechanical Properties①
- Manganese(II) and Copper(I) Compounds Based on Two Derivatives of Imidazo[1,5-a]pyridine: Synthesis,Structures, Magnetic Properties, and Catalytic Activity①