
成人起病的脑白质病伴轴索球样变和色素胶质细胞(ALSP)的影像表现
2022-04-06 06:37张晓倩刘泽宇有慧冯逢
国际医学放射学杂志 2022年2期
张晓倩 刘泽宇 有慧 冯逢
成人起病的脑白质病伴轴索球样变和色素胶质细胞(adult-onset leukoencephalopathy with axonal spheroids and pigmented glia,ALSP)是一种以染色体5q32 上集落刺激因子1 受体(colony-stimulating factor 1 receptor,CSF1R)为致病基因的罕见常染色体显性遗传病,是遗传性脑白质病的最多见亚型之一[1],1984 年由Axelsson 首次报道,自2013 年被认定为独立的疾病实体[2]。ALSP 包括遗传性弥漫性脑白质病伴轴索球样变 (hereditary diffuse leukoencephalopathy with spheroids,HDLS)和色素型正染性脑白质营养不良(pigmentary orthochromatic leukodystrophy,POLD)[3],与其他成人脑白质病有重叠表现。该病临床罕见,多表现为认知障碍、行为或精神性格改变、运动障碍,并可伴癫 、锥体束征、帕金森病样改变和步态障碍等[4]。ALSP 临床表现复杂且个体差异大,极易被误诊。目前已报道70 余种CSF1R 基因突变类型[5-6],并已有丙氨酰转移RNA合成酶2(alanyl-transferRNA synthetase 2,AARS2)等新生致病突变的报道[7],因此独立应用遗传学手段诊断ALSP 作用有限。本文总结3 例ALSP 病人的神经影像学表现、临床表现、基因及组织活检,并探讨多项检查结果联合诊断的价值,以期提高早期诊断该病的能力。
1 资料与方法
1.1 一般资料 对3 例就诊于北京协和医院神经内科并经基因遗传学或病理组织学确诊的ALSP 病人进行分析。3 例均为女性,年龄36~44 岁,平均(39±4.36)岁。起病至临床就诊时间间隔为11~19 个月,平均(14±4.36)个月。外院首诊分别诊断为“脑梗死”、“正常颅压性脑积水”和“痴呆待查”。3 例病人均有认知障碍和锥体束征,但无偏瘫或单瘫。存在精神行为异常和帕金森病样表现的有2 例;存在癫、共济失调和步态异常的有2 例;存在球麻痹症状、感觉障碍和二便失禁的仅1 例。3 例病人血清生物化学检验、抗体、炎症指标、肿瘤生物标志物、脑脊液检查等结果均为阴性。
1.2 设备与方法 采用GE Discovery MR 750w 3.0 T、GE Signa 1.5 T MRI 设备和德国西门子SOMATOM Definition 双源螺旋CT 扫描设备,扫描范围自颅底至颅顶。常规MRI 序列包括横断面T1WI、T2WI、液体衰减反转恢复(FLAIR)和扩散加权成像(DWI),生成表观扩散系数(ADC)图;对比增强检查采用对比剂钆喷酸葡胺(Gd-DTPA,浓度0.5 mol/L,北陆药业)。CT 扫描层厚为5 mm。
1.3 基因遗传学检测 所有病人及家属均签署基因遗传学检测知情同意书。从病人新鲜外周血白细胞中提取基因组DNA,在美国IlluminaHiseq 测序仪上使用“下一代”测序技术对整个外显子进行测序,并通过Sanger 测序验证。
2 结果
2.1 影像学表现 3 例病人行MRI 检查,白质病变均累及双侧侧脑室周围和深部白质,呈额、顶叶分布,可累及胼胝体;2 例可累及锥体束,均未累及脑干。3 例病变均呈不对称分布,其中2 例病灶呈大片状、融合分布,另1 例病灶呈斑片状、散在分布;3 例均未见病灶强化及皮质下U 纤维受累;除白质病变外,3 例病人均可见侧脑室扩大和病变区DWI 点片状高信号,2 例病人伴脑叶萎缩。2 例病人行CT 检查,可见微小点状钙化。详见图1-3。

图1 ALSP 病人,女,36 岁。A-D 图分别为横断面T1WI、T2WI、DWI 及ADC 图,可见双侧侧脑室旁额顶叶深部白质有斑片状T1 低信号、T2 高信号影,DWI 上呈不均匀高信号,ADC 图上可见大部分呈高信号,伴小片状低信号扩散受限区。E-G 图为横断面DWI 影像,H 图为FLAIR 影像,均可见锥体束受累征象。
2.2 基因检测结果 3 例病人中仅2 例行基因遗传学检查且均存在CSF1R 的位点突变。其中,1 例病人双亲基因筛查均无异常;另1 例的父亲存在1个位点突变而母亲未检测,病人符合杂合突变。
3 讨论
随着对ALSP 典型病理和遗传表现的认识增加,世界各地对遗传/散发病例和家系的报道也越来越多[9-11],提示该病的发病率可能被低估。ALSP 具有一定的发病特征,包括年龄、典型临床症状和影像表现,结合基因或组织病理学检测可明确诊断。
3.1 临床特点 ALSP 可发生于18~78 岁,平均发病年龄为(43±11)岁,95%的病例发病年龄<60 岁,均>10 岁[8]。女性发病年龄较男性年轻(分别为40 岁和47 岁),但男女发病率无差异[9]。ALSP 的临床特征多种多样,鉴别诊断亦涉及广泛。以双侧锥体束受损和运动障碍就诊的病人,影像表现为双侧脑室周围多发白质病变,需要与多发性硬化等脱髓鞘疾病鉴别[10-11];以认知功能减退就诊的病人,需要考虑与额颞叶变性、早发型阿尔茨海默病、血管性痴呆等疾病鉴别[12]。病人有步态异常、痴呆及尿失禁,影像学检查提示脑室扩张,需除外正常颅压性脑积水。此外,白质病变还应与中枢神经系统血管炎、小血管疾病以及其他遗传性白质脑病和白质营养不良相鉴别[13]。例如常染色体显性遗传病合并皮质下梗死和白质脑病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)可出现“2 次以上的卒中样发作”,而ALSP 病人中不常见。ALSP 最常见的临床症状是认知减退、精神症状、锥体束征和运动障碍[8,14]。在本组3 个病例中,发病年龄、疾病进展过程及临床表现与既往文献相一致,但未包括男性病例。
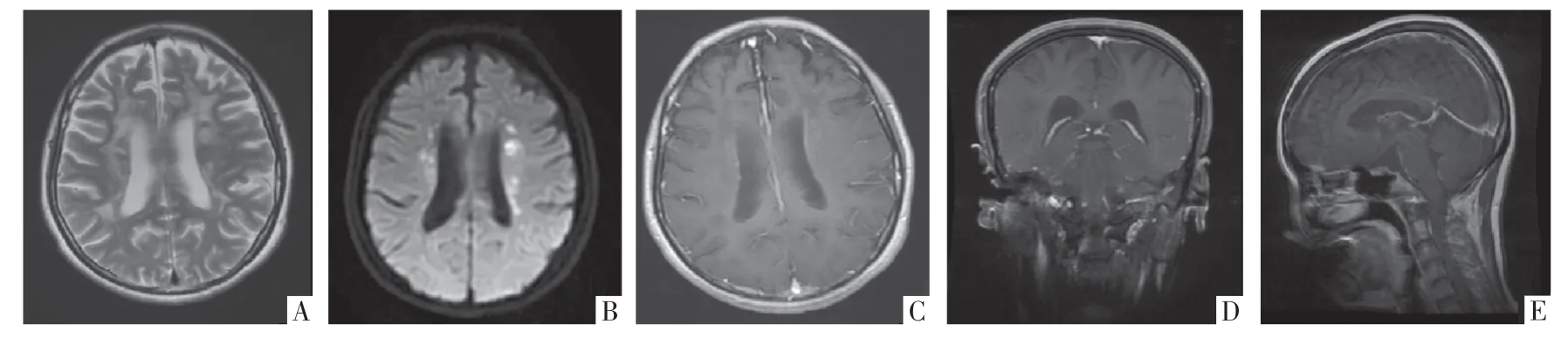
图2 ALSP 病人,女,37 岁。A-B 图分为T2WI、DWI 影像,提示双侧侧脑室旁额、顶叶深部白质T2WI 高信号,病灶内多发点片状DWI 高信号。C-E 图分别为横断面、冠状面和矢状面增强MRI 影像,病灶不伴钆剂强化。

图3 ALSP 病人,女,44 岁。A-D 图分别为横断面T1WI、T2WI、DWI、ADC 影像,双侧侧脑室旁额、顶叶深部白质可见T1 低信号、T2 高信号,伴胼胝体变薄和胼胝体压部扩散受限信号(图C、D),双侧侧脑室扩张。E 图为CT影像,左侧侧脑室旁可见点状钙化灶。
ALSP 的主要病理特征为轴索球样变和色素巨噬细胞沉着,镜下病变白质中可见轴突和髓鞘弥漫性损伤、丢失,神经轴索肿胀(球样变)和大量星形胶质细胞增生[15]。本文1 例病人经活检证实有以上组织学特点。ALSP 的典型致病突变为CSF1R,这是一种表达于小胶质细胞表面的酪氨酸激酶受体[2]。本文2 例病人证实存在CSF1R 突变,其中1 例病人父亲存在1 个位点基因突变(该病人基因检查符合杂合突变),符合基因学诊断标准。有研究[7]发现CSF1R 突变阴性的ALSP 似乎与AARS2 等基因的突变有关,其临床和影像学特征与CSF1R 型ALSP 病人相似;不同之处在于AARS2 病人可见常染色体隐性遗传模式,发病年龄相对较小(可于儿童期发病),可伴致死性心肌病,可存在卵巢功能衰竭[7,16-17]。由此可见,ALSP 具有遗传异质性。
3.2 影像学特征 2018 版CSF1R 型ALSP 诊断标准[8]提示该病于MRI 上表现为脑内多发不伴钆剂强化的白质病变,病灶在疾病早期可散在分布且不对称,但后期出现逐渐融合、弥漫、对称的分布趋势;最常见额、顶叶受累,可见颞枕叶受累;常见伴脑叶萎缩和侧脑室扩大,而大脑深部灰质核团、外囊和脑干极少受累,偶见文献报道延髓受累[18-19]。在该病早期就可观察到胼胝体变薄伴信号异常[20-21]。在多数病例中可以观察到锥体束异常信号和白质病灶扩散受限[9,21]。本组3 例ALSP 病例白质病变位置分布及信号特点,与诊断标准及既往文献报道一致,未见单独额叶受累或颞枕叶受累,1 例病灶呈额顶叶分布,额叶受累为著,考虑与病程相关。1 例病人在近50 d 的住院过程中,进行过4 次颅脑MRI 检查,均存在病灶区DWI 持续高信号,与缺血性病变不同,该表现可持续数月或更长时间,可能反映髓鞘内水肿[22-23]。
CSF1R 型ALSP 可以出现钙化,常见于两侧侧脑室前后角周围的额、顶叶白质区域,呈“踏脚石”样改变[10]。钙化灶可以很小,可使用1 mm 薄层CT 检出[5,11]。本文报道2 例病人在CT 上均可见侧脑室旁白质病灶内点状钙化。此外,根据病人的磁敏感加权成像(SWI)和磁共振波谱成像(MRS)也可辅助诊断。SWI 上可见侧脑室旁白质内散在低信号,结合幅值图和相位图可提示钙化成分[21];MRS 可存在N-乙酰天冬氨酸(NAA)峰值降低,胆碱(Cho)、乳酸(Lac)和肌醇(mI)峰值升高,且部分无症状性CSF1R基因携带者早期即可出现上述代谢改变[22,24]。
3.3 鉴别诊断 鉴于ALSP 的典型白质病变影像学表现,需要与以下遗传代谢疾病鉴别:①其他类型白质营养不良,如肾上腺脑白质营养不良,多见于男性,女性病人症状多较轻,罕见脑功能障碍,且影像学以顶枕部白质病变为主;异染性脑白质营养不良(成人型)可始于额叶白质,继而发展至脑室周围及皮质下广泛的对称性病灶,中央白质区可早期出现“虎斑样”改变。②成人型亚历山大病,以额叶白质受累为主,影像表现为病灶呈T2高信号,向颞顶叶白质和外囊延伸,可累及皮质下弓形纤维、基底节和脑干,可伴透明隔囊性扩张;随着病变的进展,可见脑白质空洞和萎缩。③CADASIL,其致病基因为NOTCH3基因,可见双侧颞极脑白质特征性T2信号升高,外囊、脑干及胼胝体常受累,伴多发的腔隙性梗死灶,皮肤活检电镜下可见嗜锇颗粒。
本病还需与DWI 高信号的病变进行鉴别,如脑血管疾病(梗死等)、感染及脱髓鞘在内的炎性疾病、原发和继发性肿瘤疾病,结合病程、临床症状、体征及增强MRI 检查等有助于鉴别。
3.4 小结 对于ALSP 病人,临床症状结合相对特异的MRI、CT 影像表现有助于缩小鉴别诊断范围,从而提示完善基因检测来明确诊断。当青壮年伴有/无精神运动障碍的进行性痴呆时,其MRI 提示额顶叶受累为著的脑白质病变,伴受累部位脑叶萎缩、胼胝体变细、DWI 上白质高信号并病灶不伴强化,CT 提示微小钙化时,应当考虑到ALSP 的诊断。结合典型的组织学改变及基因检测,可确诊该病。
猜你喜欢
锦州医科大学报(2022年3期)2022-06-06
东南大学学报(医学版)(2021年6期)2022-01-27
影像研究与医学应用(2021年15期)2021-09-12
昆明医科大学学报(2021年3期)2021-07-22
中国CT和MRI杂志(2020年2期)2020-03-10
医学新知(2019年4期)2020-01-02
保健与生活(2019年15期)2019-09-12
中西医结合心血管病电子杂志(2016年23期)2017-03-03
医学综述(2015年22期)2015-12-09
安徽农业科学(2015年10期)2015-04-24
