
肝内和肝外胆管癌的分子改变及靶向治疗
2022-03-29 10:31:08王英珍曾也婷王心睿黄雄飞
临床与实验病理学杂志 2022年1期
王英珍,曾也婷,王心睿,黄雄飞
胆管癌(cholangiocarcinoma,CCA)起源于胆管上皮细胞,是仅次于肝细胞癌的常见原发性肝脏恶性肿瘤[1]。根据解剖部位分为两种亚型:肝内胆管癌(intrahepatic cholangiocarcinoma, ICC)和肝外胆管癌(extrahepatic cholangiocarcinoma, ECC)。ECC又可分为肝门周围胆管癌(perihilar cholangiocarcinoma, pCCA)和远端胆管癌(distal cholangiocarcinoma, dCCA)[2]。全球CCA发病率近十年持续增加[3],尤以ICC最为显著[4]。中国CCA患者约占全球CCA患者的55%[5]。因此,我国深入研究CCA具有重要意义。
过去较少根据分子改变对CCA患者进行精确分组,这可能是其高病死率的原因之一。目前国内外研究相继发现了多个CCA靶点,分子靶向治疗有望成为CCA治疗的新的突破点。美国FDA已经批准部分靶向药物上市,还有一些药物正处于临床试验阶段(表1)。ICC的主要分子改变有IDH1、FGFR、KRAS以及染色质重塑;ECC的主要分子改变有KRAS、EGFR/HER2、PRKACA和PRKACB、ELF3[6]。本文分别对ICC和ECC两种亚型相应的分子改变及靶向治疗进行论述(图1)。
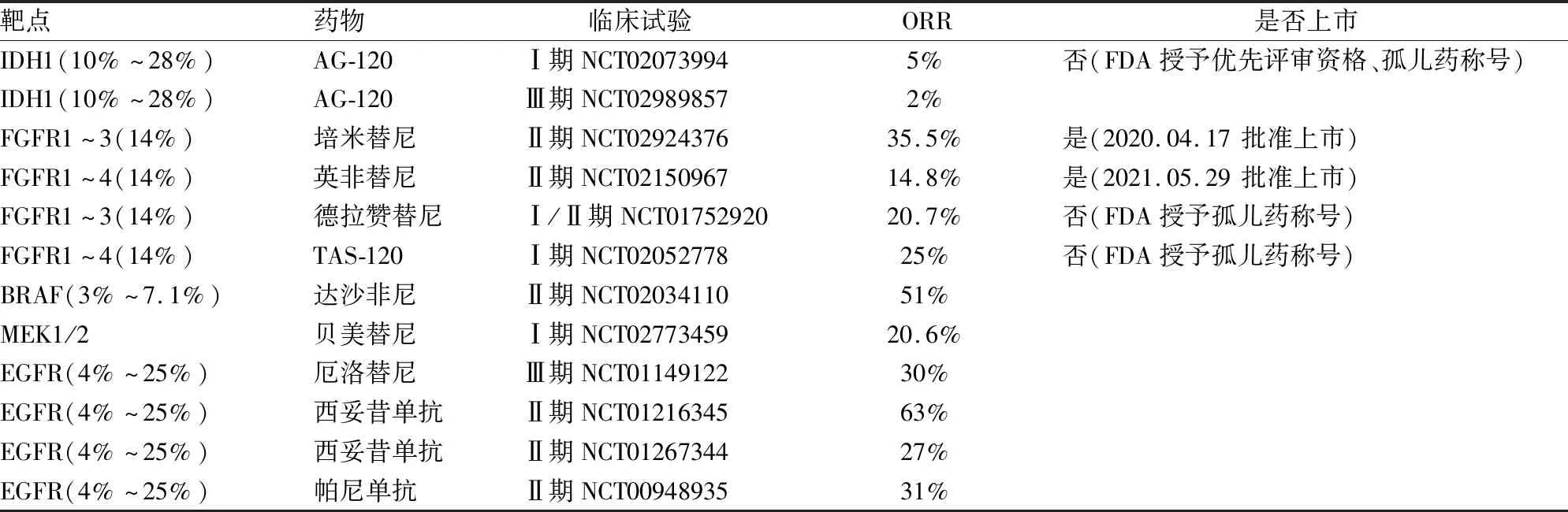
表1 CCA的主要靶向药物及上市情况
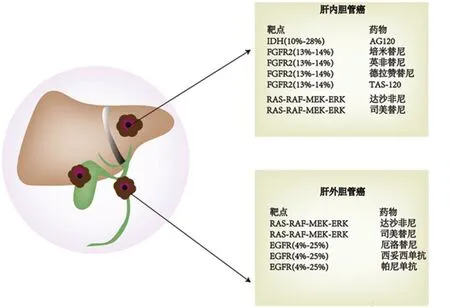
图1 ICC和ECC的分子改变及主要靶向药物
1 ICC的分子改变及靶向药物
1.1 IDH突变及其靶向药物10%~28%的ICC发生异柠檬酸脱氢酶(IDH)突变[7],IDH1 R132是最常见的突变位点[8]。IDH是三羧酸循环的关键酶,生理情况下催化异柠檬酸氧化脱羧生成α-酮戊二酸(α-KG)[9]。当IDH结构域发生错义突变时[10],大量肿瘤代谢物2-羟基戊二酸(2-HG)在细胞中蓄积[11]。2-HG竞争性地抑制以α-KG为辅因子的双加氧酶活性,引起表观遗传改变,如DNA甲基化增加,从而使基因表达失调[12]。
艾伏尼布(Ivosidenib, AG-120)是针对IDH1突变(mIDH1)的靶向抑制剂。Ⅰ期试验(NCT02073994)评估了艾伏尼布对于mIDH1晚期CCA患者的安全性及活性,5%患者实现部分缓解(partial response, PR),中位无进展生存期(progression free survival, PFS)为3.8个月,中位总生存期(overall survival, OS)为13.8个月[13]。Ⅲ期试验(NCT02989857)评估该药对比安慰剂治疗一线化疗失败的晚期mIDH1 CCA患者[14],艾伏尼布组客观缓解率(objective response rate, ORR)为2%,安慰剂组病例均未实现客观缓解。中位PFS(2.7个月vs1.4 个月)和中位OS(10.8个月vs6.0个月)均有改善。IDH2抑制剂恩西地平(AG221),已经在mIDH2实体瘤(包括ICC)中进行临床试验(NCT02273739),结果尚未披露[15]。
1.2 FGFR基因融合及其靶向药物成纤维细胞生长因子受体(FGFR)家族由FGFR1~5五种受体组成。该家族信号转导失调与ICC密切相关,13%~14%的ICC发生FGFR2易位/融合[16],FGFR2-BICC1是最常见的融合形式[16]。易位/融合首先通过激活FGFR受体,进而活化下游Ras-Raf-MEK、JAK-STAT或PI3K-AKT-mTOR等信号通路,驱动肿瘤形成[17]。
美国FDA已经批准培米替尼(Pemigatinib)和英非替尼(Infigratinib,BGJ398)上市。培米替尼的Ⅱ期实验结果(NCT02924376)显示,FGFR2融合或重排患者ORR为35.5%,疾病控制率(disease control rate, DCR)为82%,中位OS为21.1个月[18]。英非替尼的Ⅱ期实验结果(NCT02150967)显示,ORR为14.8%,DCR为75.4%,且FGFR2融合患者效果更好,ORR为18.8%,DCR为83.3%[19]。德拉赞替尼(Derazantinib, ARQ087)治疗FGFR2融合但无法行手术切除的ICC成年患者的Ⅰ/Ⅱ期试验(NCT01752920)结果显示,ORR为20.7%,DCR为82.8%,中位PFS为5.7个月[20]。Ⅰ期TAS-120(Futibatinib)试验(NCT02052778)中,28例FGFR基因融合的ICC患者,ORR达25%,DCR达78.6%[21-22]。总之,FGFR的靶向药物治疗效果均较理想。
1.3 染色质重塑基因失活及其靶向药物ICC频繁发生染色质重塑复合物失活[23],该复合物由ARID1A、BAP1和PBRM1等抑癌基因组成[6]。BAP1编码染色质重塑的核去泛素化酶,ARID1A和PBRM1是染色质重塑复合物的亚基[23]。Churi等[24]研究结果显示, 分别有20%、9%和11%的ICC存在ARID1A、BAP1和PBRM1的失活。BAP1通过抑制SLC7A11(胱氨酸转运体)和刺激铁死亡以限制肿瘤生长[25]。PBRM1和ARID1A通过调控错配修复以实现以对DNA损伤的调节[26]。
针对染色质重塑复合物抑制剂的临床研究较少,包括两类,一类是组蛋白去乙酰化酶(HDAC)抑制剂:伏立诺他(Vorinostat)、罗米地辛(Romidepsin)和丙戊酸;另一类是DNA甲基转移酶(DNMT)抑制剂:阿扎胞苷(Azacitidine)和地西他滨(Decitabine)[27]。在一项丙戊酸联合S-1(5-氟尿嘧啶的衍生物)治疗12例晚期胰胆管癌患者的Ⅰ/Ⅱ期临床研究中,仅1例患者疾病进展,证明联合治疗具有一定的效果[28]。
1.4 BRAF、KRAS突变及其靶向药物ICC常发生RAS-RAF-MEK-ERK信号通路异常表达, 其中KRAS和BRAF在该通路中起重要作用,8.6%~24.2%的ICC发生 KRAS 突变[29],3%~7.1%的ICC发生BRAF突变(BRAF V600E突变最常见)[29],通过与细胞周期调节蛋白相互作用影响细胞周期,并干预促凋亡蛋白Bax信号转导以逃避细胞凋亡。
目前尚无KRAS突变的直接有效抑制剂,治疗方案主要是抑制KRAS的下游蛋白。维罗非尼(Vemurafenib)和达沙非尼(Dabrafenib)是BRAF V600E的靶向抑制剂。司美替尼(Selumetinib)、曲美替尼(Trametinib)以及贝美替尼(Binimetinib)是MEK1/2的靶向抑制剂。试验证明组合用药效果更好,在达沙非尼与曲美替尼联合治疗BRAF V600E突变CCA患者的Ⅱ期试验中(NCT02034110),总反应率达51%,中位PFS为9个月,中位OS为14个月[30],与一线CisGem化疗方案疗效相当。司美替尼联合CisGem化疗(NCT01242605)治疗晚期CCA的中位PFS为6.4个月,在8例可评估的患者中,3例实现了PR,5例疾病稳定[31]。在Ⅰb期试验(NCT02773459)中,贝美替尼联合卡培他滨治疗一线化疗失败的CCA患者,中位PFS和中位OS达4.1个月和7.8个月,ORR和DCR达20.6%和76.5%[32]。
2 ECC的分子改变及靶向药物
2.1 BRAF、KRAS突变及靶向抑制剂ECC比ICC存在更高频率的RAS-RAF-MEK-ERK信号通路失调,有研究表明12%~40%的ECC存在KRAS突变,ECC很少发生BRAF突变[33]。靶向抑制剂与ICC相同,故不再赘述。
2.2 EGFR/HER2过表达及其靶向药物ERBB受体酪氨酸激酶家族包括4个成员:ERBB1(EGFR)、HER2、ERBB3及ERBB4[34]。4%~25%的ECC过表达EGFR/HER2[33],通过胞外偶联表皮生长因子、肝素结合因子等,激活胞内JAK/STAT、RAS/MEK/ERK或PI3K/AKT信号通路,促进CCA细胞增殖侵袭[35]和肿瘤基质血管生长。
以HER2为靶点的抑制剂包括曲妥珠单抗、帕妥珠单抗和拉帕替尼(Lapatinib)。以EGFR为靶点的药物有两类:一类是通过胞内途径起作用的小分子酪氨酸激酶抑制剂,如厄洛替尼(Erlotinib)、吉非替尼(Gefitinib)和阿法替尼(Afatinib);一类是通过胞外途径起作用的单克隆抗体,如西妥昔单抗(Cetuximab)和帕尼单抗(Panitumumab)[2]。厄洛替尼联合GEMOX化疗(吉西他滨+奥沙利铂)对比单独GEMOX化疗治疗晚期CCA的Ⅲ期临床试验(NCT01149122),结果显示,联合化疗组ORR明显改善(30%vs16.0%,P=0.005),但患者总生存期没有延长,中位OS均为9.5个月[36]。西妥昔单抗联合GEMOX化疗治疗不可切除的晚期或转移性CCA的Ⅱ期试验(NCT01216345),结果显示,ORR达63%,9例患者在治疗后成功接受了降期手术[37]。西妥昔单抗联合GEMOX化疗对比单独GEMOX化疗治疗KRAS突变的晚期CCA患者的Ⅱ期试验(NCT01267344),结果显示,尽管联合化疗组PFS有改善的趋势(6.7个月vs4.1个月,P=0.05),但ORR(27%vs15%,P=0.120)和OS均无改善(10.6个月vs9.8个月,P=0.910)[38]。帕尼单抗联合吉西他滨和伊立替康治疗晚期CCA的Ⅱ期试验(NCT00948935),结果显示,主要终点5个月无进展生存率达69%,次要终点ORR和中位OS达31%和12.9个月[39]。针对该靶点的联合治疗对比单独化疗的疗效尚无统一定论,但仍给CCA患者的治疗提供了希望。
2.3 PKA(PRKACA和PRKACB)基因融合及其靶向药物PRKACA和PRKACB是PKA的催化基团,10%的ECC频繁发生该基因融合[6]。Nakumura等[6]在ECC中检测到ATP1B-PRKACA和ATP1B-PRKACB基因融合,这些融合增强了PRKACA和PRKACB的表达,进而激活下游MAPK信号通路。
PRKAR1A是PKA的调节亚基,在CCA细胞中过表达[40]。沉默PRKAR1A能够诱导CCA细胞生长抑制和凋亡,提示PRKAR1A可能作为CCA的分子治疗靶点[40]。特异性PKA抑制剂异喹啉H89(Isoquinoline H89)和cAMP类似物8-氯-环磷酸腺苷(8-Cl cAMP)、8-溴-环磷酸腺苷(8-Br cAMP),能够显著抑制CCA细胞生长[40]。
2.4 ELF3缺失Nakamura等[6]对260例胆道肿瘤患者进行了全面的外显子和转录组测序分析,发现9.5%的ECC存在ELF3基因突变,这是ECC的一个新驱动基因。上皮特异性ETS样转录因子(ELF3),能够增强多种抑癌基因转录激活[41],如转化生长因子2型β受体(TGFBR2)和表皮生长因子1(EGF1)[42]。在另外一个基因组分析中,Yachida等[43]对60例胆道肿瘤进行外显子序列分析,结果显示13.3%的ECC存在ELF3突变。随后的研究建立了敲除ELF3的胆道肿瘤细胞模型,与对照组相比ELF3敲除细胞的侵袭和迁移能力均显著增加。因此,推断ELF3可能在ECC中起到肿瘤抑制作用,提示ELF3可作为治疗CCA的一个靶点。目前尚无针对该靶点的靶向药物。
3 结语
CCA靶向药物的研发仍然需要克服诸多问题,如可供研究的患者病例数少、分子改变异质性大、后期易引发耐药等。未来的研究应持续专注于靶点的识别及靶向药物的开发,以使靶向治疗能够造福于更多CCA患者。此外,还需要尽可能地探索靶向治疗与其它治疗方法的联合使用,如免疫治疗、化疗等,以期找到更佳的治疗方式,从而改善CCA患者的预后。
猜你喜欢
Journal of Hainan Medical College(2023年15期)2023-12-06 07:54:42
时代英语·高三(2022年3期)2022-11-10 01:20:51
中老年保健(2021年3期)2021-12-03 02:32:25
大电机技术(2021年3期)2021-07-16 05:38:34
中国生殖健康(2020年7期)2020-12-10 07:48:51
皮肤性病诊疗学杂志(2020年4期)2020-09-02 07:36:58
中外文摘(2020年13期)2020-08-01 01:07:06
时代英语·高三(2019年4期)2019-09-03 02:11:40
中国卫生标准管理(2015年16期)2016-01-20 09:26:32
医学研究杂志(2015年7期)2015-06-22 11:01:36
