
复杂微球缓释制剂研发项目管理方法
2022-03-23 07:43:54樊素然艾尔菲丁阿尼娃尔
化工管理 2022年7期
樊素然,艾尔菲丁·阿尼娃尔
(1.中国人民大学商学院,北京 100872;2.新疆医科大学药学院,新疆 乌鲁木齐 830017)
0 引言
微球通常是指粒径在5~250 μm的球体,将药物溶解或分散在高分子材料基质中形成微球称为缓释微球。微球制剂通常使用大分子可降解的聚合物为骨架,给药后可逐渐降解并释放药物,有效避免血药浓度的峰谷现象、减少治疗周期内的给药剂量和给药次数,从而减小毒副作用、提高药物在体内的生物利用度以及病人的顺应性。通过微球制剂研发的新型给药系统目前正日益成为医药行业尤其是大型医药研发企业的关注焦点[1]。
局部麻醉药(局麻药)通常为小分子药物,这类药物能够在用药局部可逆性地阻断感觉神经冲动发生与传导的药物,临床期望对于手术后急性疼痛的镇痛时间一般为几天甚至一周。由于单次局麻药物的持续阵痛时间较短,导致难以满足临床期望的阵痛时间,因此常通过频繁重复给药、导管体内植入、自控式镇痛泵(PCA)以及神经破坏术等方式来延长镇痛时间。频繁重复的给药次数给患者带来很多不便,而且可能导致药物蓄积,造成中枢系统及心血管毒性。因此针对临床当前关于局麻药的使用现状,十分有必要开发一种可长时间持续阵痛的局麻药缓释制剂[2]。
如上所述,将微球缓释制剂的制备技术应用于局麻药物,既可以提高局麻药物的生物利用度、延长药物的作用时间,又可以减少给药次数,为患者带来便利。因此,早在20世纪90年代,已有将微球作为局麻药载体的报道和研究;目前,国内外已有大量的科研人员以及药物公司在进行局麻药物缓释制剂相关工作的研究。而复杂微球注射剂不同于传统的药物制剂,其研发中涉及的关键质量属性、关键物料参数、关键过程参数以及注册审批中的因素较多,所以需在整个开发流程中,通过先进的项目管理理念进行指导监控,才能尽量降低项目研发失败风险。
本文以局麻药缓释微球项目的开发为例,总结了项目管理在项目开发过程中的影响以及作用,并探讨复杂微球缓释注射剂研发中项目管理方法的应用和必要性。
1 微球缓释注射剂研发项目的特征与项目管理现状
与传统的注射剂类项目研发不同,微球缓释注射剂研发项目的整个管理过程中,涉及的人员、机构复杂,研发、临床试验、注册审批等过程中的不确定因素很多,加之微球本身性质的影响因素众多,这些都使得研发项目管理过程中的可控性难度增加。
目前,虽然微球缓释注射剂的研发面临种种挑战和风险,但由于其特有的临床优势,已有部分商品化的微球制剂获批上市。较为代表性的公司有辉瑞、阿斯利康、益普生、辉凌等制药公司。涉及的活性成分如:醋酸亮丙瑞林、醋酸曲普瑞林、艾塞那肽、利培酮、生长激素等;治疗领域分布在前列腺癌、子宫内膜异位症、II型糖尿病、抗精神病类、儿童生长激素缺乏症等。
由于可注射缓释微球技术的巨大市场潜力,除了以上已上市品种,世界各国的制药企业和研发机构正在研究多种药物的缓释制剂,如促红细胞生成素(EPO)、γ-干扰素(γ-IFN)、人粒细胞-巨噬细胞集落刺激因子(GM-CSF)、白介素(IL)-1α等。
微球缓释注射剂研发过程的项目管理,既具有一般项目管理的特点,也具有其独特之处。
1.1 安全性和法规要求
由于可生物降解聚合物材料在人体内逐渐降解后的代谢产物可被人体吸收,因此被广泛应用于微球药物中。微球制剂影响因素众多,难于仿制,微球缓释制剂开发多以2.2类改良型新药的方式进行申报;我国于2020年6月出台了《化学药品改良型新药临床试验技术指导原则(征求意见稿)》,提出改良型新药应具有明显的临床优势。
1.2 专业要求高
与天然的聚合物相比,通过人为控制聚合制备工艺,可使化学合成的聚合物材料达到药用辅料级别;改变聚合物粘度、分子量等关键参数,可以自由地控制载药微球的降解速度。但是,聚合物对载药微球性质的影响,会因为药物本身的亲疏水性和特殊官能团的不同而显示出不同的影响结果,微球药物的研发需要有专业的技术背景和高水平的技术平台[3]。
1.3 复杂程度高
微球制剂的研发影响因素诸多。长效缓释制剂的设计开发,需考虑到诸多方面的影响,如产品质量属性涉及到的无菌、可注射性、延长药物释放、生物相容性等[4-7]。Francesca Selmin 等研究者在公开发布的文献中也提到了产品属性[8](target product prof ile, TPP)在长效植入制剂研发中发挥的重要作用,如图1所示。
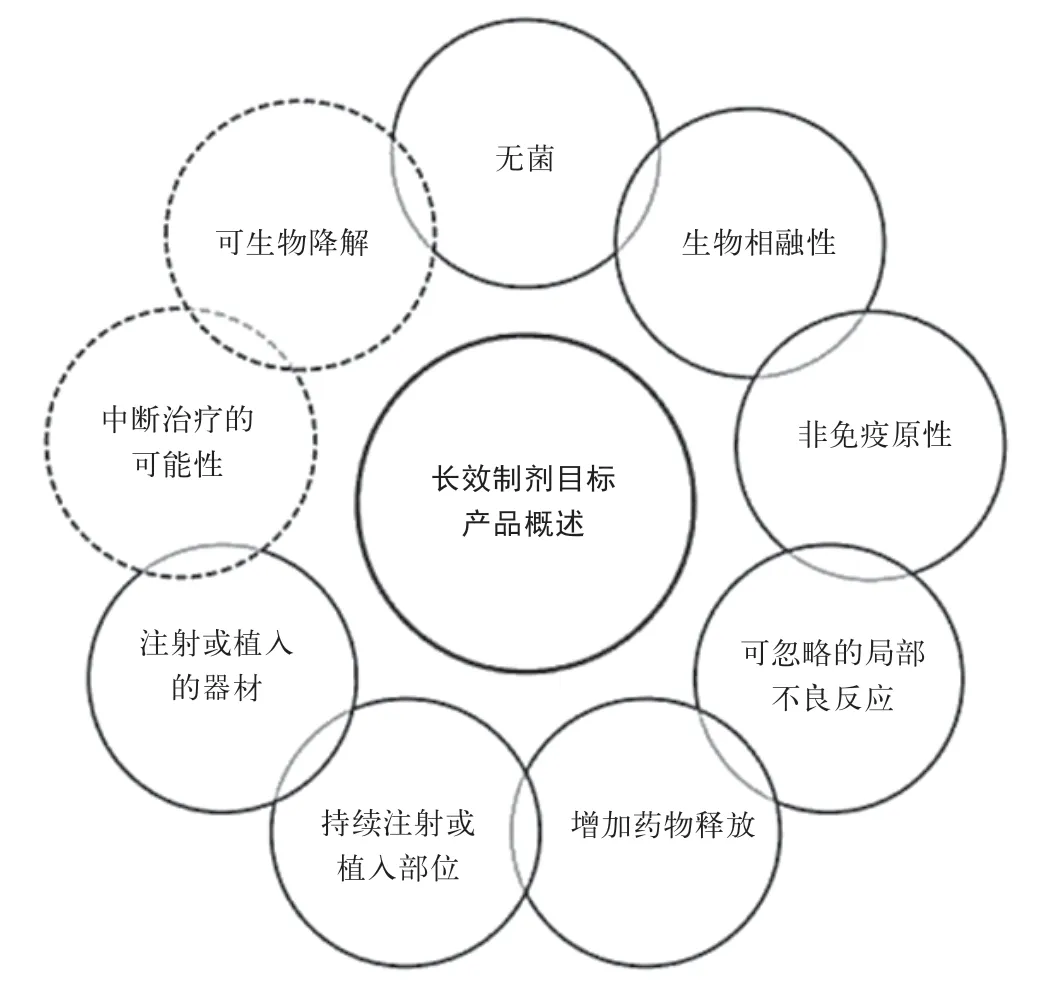
图1 长效植入剂的TPP
1.4 研发周期长
就药物研发本身而言,从实验室研发、小试、中试到产量化上市过程中,需要很多的实验、测试等,同时还需要注册申报、临床试验以及注册上市后的的跟踪监督。结合微球研发本身的复杂性,导致微球药物研发的周期较长。绿叶制药作为我国首屈一指的高端制剂研发企业,在特殊制剂研发领域深耕多年,近日,其自主研发的瑞新托(注射用利培酮微球)获中国国家药监局批准上市,研发周期历时近10年,这已是行业翘楚。
1.5 失败风险高,难以仿制
以微球为主的新型释药系统开发,由于存在较高的技术壁垒,一些关键技术多掌握在欧美等发达国家,研发中既需要有专业的技术平台,也要有特殊的生产线,加之特殊制剂对生产条件的要求比较严格,导致微球药物研发的失败风险极高,难以仿制。首例多肽类微球制剂是20世纪80年代上市的醋酸曲普瑞林缓释微球,上市几十年来,仍然无仿制产品上市;就微球生产的设备而言,以膜乳化工艺为例,其所使用的设备,在欧美和日本均已上市多年,而国内的设备生产才刚刚起步,生产商屈指可数,设备质量有待改进。
2 微球缓释注射剂研发项目管理阶段
美国项目管理协会的项目管理知识体系指南中指出了项目的基本概念,强调项目是一个临时性的工作,它所创造的产品、服务或成果是独特的。项目管理即是要将实施过程中的管理知识、技能、技术、工具应用在执行活动当中,使其满足最终的要求;通过合理的运用和整合,使得项目管理过程得以实现;从而使组织能够有效地执行项目计划,推动项目高效的进行。项目生命周期是通过一系列的项目管理活动进行的,即项目管理过程。各个项目管理过程通过合适的项目管理工具和技术将一个或多个输入转化成一个或多个输出,输出可以是交付成果或结果。项目管理过程组即对项目管理过程进行逻辑分组,以达成项目的特定目标。过程组不同于项目阶段,项目管理过程可分为以下五个项目管理过程组,即启动过程组、规划过程组、执行过程组、监控过程组、收尾过程组。此外,过程还可以按知识领域进行分类,所谓的知识领域是指用项目管理过程中所需要的知识内容来区分的不同内容和领域,并用其所包含的输入、输出、过程、实践、技术和工具进行描述,各个知识领域相互联系,大部分项目通常使用十个知识领域,包括:项目的整合管理、项目的范围管理、项目的进度管理、项目的成本管理、项目的质量管理、项目的资源管理、项目的沟通管理、项目的风险管理、项目的采购管理、项目相关方的管理。以上介绍的项目管理所涉及到的五个项目管理过程组和十个知识领域,也被称为“十五矩阵”。
鉴于微球药物制剂研发的较多困难和不确定性,因此不仅需要有一套完整的项目管理方法对项目的启动、规划、执行、控制、收尾全过程进行管控,而且需要根据研发不同阶段的特点进行管理。同时质量源于设计(quality by design, QBD)方法的引入也是必不可少的,在国际人用药品注册技术协调会(ICH)质量管理体系中提出为了使质量控制保持在理想状态,一定要从药物研发和质量源于设计、质量风险管理以及药物质量体系几个方面入手。QBD理念贯穿于药品整个生命周期,对药品从研发到上市、退市的整个生命周期等进行系统、规范化的管理。质量源于设计强调把研发前已经设定的目标产品质量特性作为起点,在全面分析关键物质属性基础上,通过试验设计,研究产品的关键质量属性,确立关键工艺参数,即根据QBD的概念,药品从研发之初就已经开始考虑最终产品的质量了。以局麻药微球缓释制剂的研发为例,项目研发的全生命周期管理过程如图2所示。
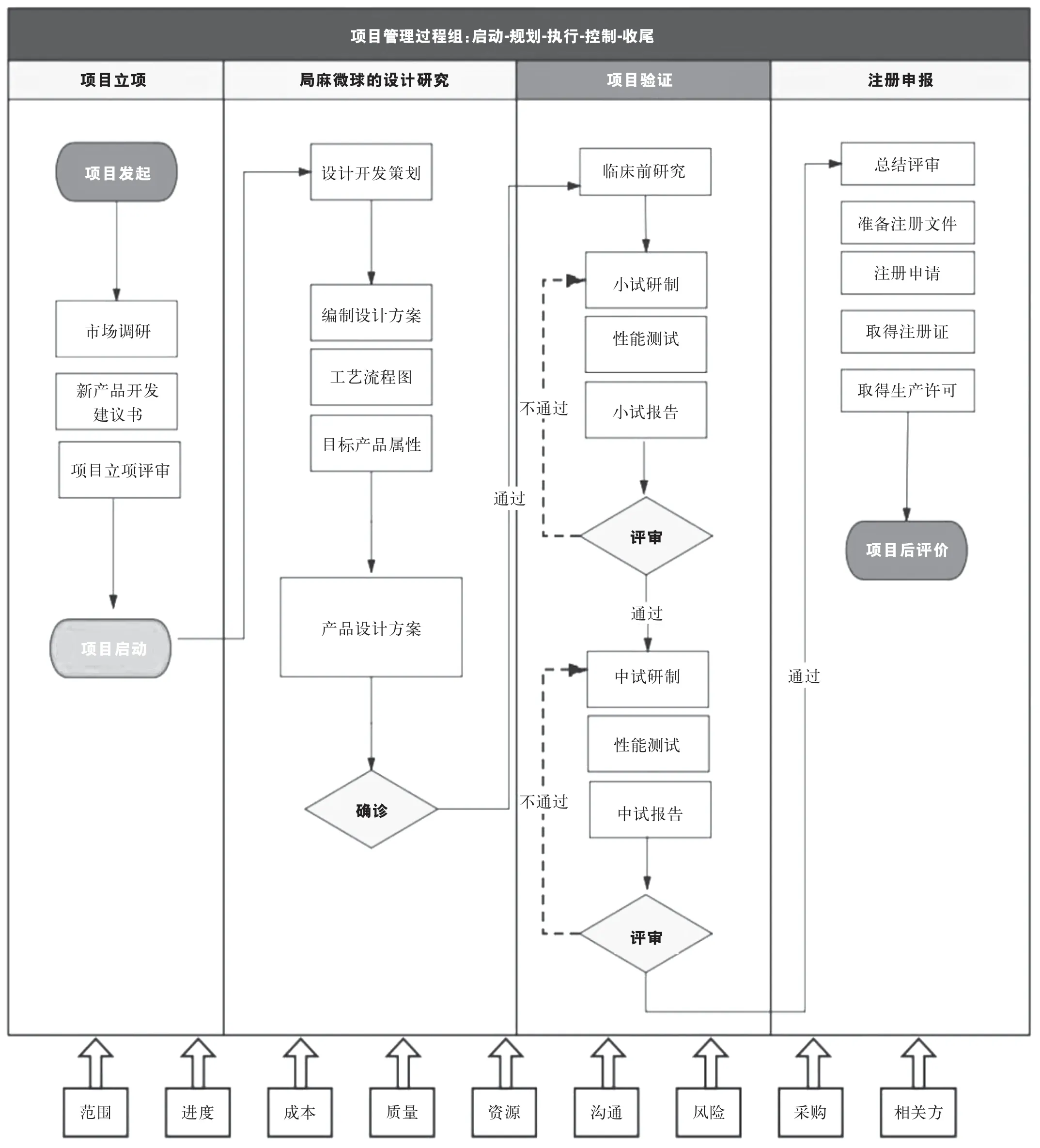
图2 局麻药缓释微球研发项目全生命周期管理
微球药物的上市品种较少,难以仿制;企业较多以505B(2)改良型新药的方式进行微球等特殊制剂的开发,即在已知活性成分的基础上,对药物的结构或者药品的处方工艺、给药途径、剂型、适应症等进行优化,开发出明显具有临床优势的药品[9]。在研发各阶段的主要内容和特点如下。
建议招标评分中,如果按100分计,报价得分不能超过30分,实施方案(包括质量保证措施、进度保证措施、资金保证措施、技术工艺措施等)为50分左右,企业资信等不能超过10分,投标亮点等 10分(投标亮点可公开竞讲,让企业互相有所评判,找出各自不足)。评分时要聘请资深专家,可随时就方案对企业进行质询。
2.1 项目立项阶段
项目立项阶段,包括项目的开起、市场调研、开发建议书、立项评审等。通过立项,在项目开始时就认真考虑和调研了项目的商业论证、项目效益和相关方,确保项目具有可行性、符合组织的战略目标,相关方达成共识。
该阶段输出的主要交付成果是初步的目标产品概况,从整体上明确所开发的目标产品方向。《药品管理法》第二章第十六条规定国家支持以临床价值为导向的新药研制,鼓励和推动药品技术的进步,可见缓释微球技术应用于局麻药物的制剂开发符合法规政策的鼓励。根据《药品注册管理办法》对注册分类的划分,2类新药,即境内外均为上市的创新药,指在已知活性成分的基础上,对其结构、类型、给药途径、处方工艺、适应症等进行优化,且具有明显的临床优势。以局麻微球制剂的研究项目为例,该项目在前期经过多方论证,通过启动大会,项目主持方、投资方、协调管理方达成一致意见,根据微球药物研发的特点和局麻药在临床使用中的问题,确定通过剂型改良,研发新型的缓释微球制剂。因此按照2类新药进行申报,初步确定项目各个阶段关口的时间节点和评价标准,项目正式启动。
2.2 局麻药微球缓释制剂的设计研究阶段
该阶段是项目研发的技术核心阶段,需要通过策划保证对研发过程有适当的控制,从而确保产品质量目标得以实现。通过专业技术的深入研究,确立产品和原材料的质量要求。
根据前期论证,该阶段确定了所开发的局麻药微球的TPP(target product prof ile):从阶段性来说,主要分为临床(clinical)、市场(marketing)、药物产品(drug product)、药物(drug substance)几个方面,设置不同的考察因素(TPP elements)以及最优(preferred)、最低(minimum)限度,并明确关键指标项目(CQA category)。
对于局麻药缓释微球的开发,在实验室的初期研究阶段,通过工艺预筛选确定采用水包油型的单乳法包裹疏水的小分子药物,工艺流程如图 3所示。

图3 水包油单乳法制备局麻药缓释微球的工艺流程
GMP(good manufacturing practices)中指出质量风险管理是在整个产品生命周期中采用前瞻或回顾的方式,对质量风险进行评估、控制、沟通、审核的过程。风险评估是一种有价值的基于科学的分析过程,将该技术用于质量风险管理,有助于确定对产品关键质量属性产生影响的工艺参数和物料属性。在传统的质量管理中,质量风险管理的评估和管理多采用非正式的方法,以风险分析人员的经验和知识来进行,随着统计分析工具和新方法的开发,一些科学的风险管理工具被应用。在局麻药缓释微球的开发设计前期,开发团队以石川(Fishbone)图为工具,确定可能对所需质量属性产生影响的潜在变量,如图4所示。

图4 局麻药微球制剂风险分析
在设计研究部分主要交付成果是产品的方案设计,包括设计开发策划、评审计划、方案汇总报告、工艺流程图、初期的产品质量标准和原辅料质量要求等。
2.3 项目验证阶段
在项目的验证阶段,通过逐级放大,依次进行小试样品的研制和评审、中试放大的验证和评审、临床前评审、临床研究几个阶段。
2.4 注册报批阶段
安全性、有效性和质量可控性是药品所必须具有的三大特性。药事管理部门对药品的审批的基础在于药品是否具备了这三大特性。安全性一方面要求药品必须在GMP条件下生产;另一方面必须控制药品对人体的副作用。产品本身处方的设计和生产过程中使用辅助制剂残留都有可能成为造成对人体副作用的原因。因此药事管理部门对安全性的审批包括审核临床前及临床数据中的安全性评价,审核药品化学指标的检测,确认生产过程是否使用对人体有较大危害的物质,有关物质含量得到控制,审核稳定性数据,确认药品在有效期内未发生超出质量标准的不利变化,工艺验证,现场检查等。
美国FDA的药品注册管理法规体系涵盖了较为全面的内容,从技术研究、申报程序、药品上市管理评价、到社会政治经济不同的纬度,提出了明确的要求和具体的管理办法。FDA对药品的监管是针对整个药品生命周期各个环节的管理,即从药品的研发、上市到退出市场的整个过程的管理和监控:在研发阶段,通过出台一系列的指导原则,对药品的研发过程给予科学性、规范性的建议和指导;在生产、销售等环节,通过实施GXP进行监管,即新药上市和上市后的管理都是强制性。为保证药品全生命周期监管的有效性,FDA针对申请上市产品的不同环节,制定了明确的法规要求,就政府部门的管理范围、职能等进行了明确。
在药品生命周期的不同阶段,我国的《药品注册管理办法》有明确的规定,注册申请人需按程序和法定要求向国家药监部门提出临床试验、上市许可等申请。国务院药品监督管理部门应对申请注册的药品组织技术审评,对申报药品的安全、有效和质量可控、申请人的风险防控、质量管理、责任赔偿等能力进行审查;符合相关条件的才可予以审核通过和颁发注册证书。
该局麻药微球项目目前已完成临床前的研究阶段,已进入临床评审阶段,注册报批阶段需要交付的成果包括局麻药微球制剂注册申请书、注册证、产品说明书、产品标准、检测报告、质量手册等。
3 结语
通过局麻药微球缓释制剂的开发过程,总结出了一条特殊制剂研发的清晰管理框架,通过系统梳理和总结项目研发的每个过程包含的具体工作和要求,明确了各个阶段的交付成果和关键属性信息,从而提高项目实施过程的药物产品稳定性和质量可控性,以期达到提高研发成功率和降低研发周期的目的。
猜你喜欢
中华养生保健(2020年9期)2021-01-18 03:11:56
今日农业(2020年18期)2020-12-14 19:08:44
潍坊学院学报(2020年6期)2020-11-22 08:04:10
少年博览·小学低年级(2020年5期)2020-06-23 09:29:30
意林(2019年14期)2019-07-25 17:49:24
喜剧世界(2017年14期)2017-12-01 04:47:17
中成药(2017年4期)2017-05-17 06:09:48
大连工业大学学报(2015年4期)2015-12-11 04:06:50
中国当代医药(2015年29期)2015-03-01 02:07:41
中国中医药现代远程教育(2014年11期)2014-08-08 13:23:44
