
Obstructive sleep apnea aggravates neuroinflammation and pyroptosis in early brain injury followingsubarachnoid hemorrhage via ASC/HIF-1α pathway
2022-03-19 03:31:04JunXuQianLiChenYuXuShanMaoJiaJiaJinWeiGuYingShiChunFangZouLiangYe
中国神经再生研究(英文版) 2022年11期
Jun Xu , Qian Li , Chen-Yu Xu Shan Mao Jia-Jia Jin Wei Gu Ying Shi ,Chun-Fang Zou , Liang Ye
Abstract Obstructive sleep apnea can worsen the prognosis of subarachnoid hemorrhage.However, the underlying mechanism remains unclear.In this study, we established a mouse model of subarachnoid hemorrhage using the endovascular perforation method and exposed the mice to intermittent hypoxia for 8 hours daily for 2 consecutive days to simulate sleep apnea.We found that sleep apnea aggravated brain edema, increased hippocampal neuron apoptosis,and worsened neurological function in this mouse model of subarachnoid hemorrhage.Then, we established an in vitro HT-22 cell model of hemin-induced subarachnoid hemorrhage/intermittent hypoxia and found that the cells died, and lactate dehydrogenase release increased, after 48 hours.We further investigated the underlying mechanism and found that sleep apnea increased the expression of hippocampal neuroinflammatory factors interleukin-1β,interleukin-18, interleukin-6, nuclear factor κB, pyroptosis-related protein caspase-1, pro-caspase-1, and NLRP3, promoted the proliferation of astrocytes,and increased the expression of hypoxia-inducible factor 1α and apoptosis-associated speck-like protein containing a CARD, which are the key proteins in the hypoxia-inducible factor 1α/apoptosis-associated speck-like protein containing a CARD signaling pathway.We also found that knockdown of hypoxia-inducible factor 1α expression in vitro greatly reduced the damage to HY22 cells.These findings suggest that sleep apnea aggravates early brain injury after subarachnoid hemorrhage by aggravating neuroinflammation and pyroptosis, at least in part through the hypoxia-inducible factor 1α/apoptosis-associated speck-like protein containing a CARD signaling pathway.
Key Words: apoptosis associated speck like protein containing a CARD; early brain injury; hypoxia-inducible factor 1α; nucleotide-binding domain and leucinerich repeat protein 3; obstructive sleep apnea; pyroptosis; neuroinflammation; subarachnoid hemorrhage
Introduction
Subarachnoid hemorrhage (SAH) is a very common event that occurs
frequently and is associated with high rates of morbidity and mortality,particularly in older patients.The incidence of SAH is 9.1 out of 100,000 persons annually worldwide (95% CI 8.8-9.5), and is higher in Finland, Japan,and China (Ingall et al., 2000; de Rooij et al., 2007; Macdonald and Schweizer,2017).Previous studies had reported that the short-term mortality rate for SAH ranges between 8.3% and 66.7%, with 8.3% of patients dying before hospital admission, and a survival rate ranging from 36% to 55% (Komotar et al., 2009; Chen et al., 2016b).Most SAH survivors suffer from a long-term emotional impairment, cognitive dysfunction, loss of smell and hearing, and decreased quality of life after surgery (Nieuwkamp et al., 2009).Thus, SAH can impose a substantial burden on the patient’s family and on society.Increasing evidence shows that early brain injury (EBI, defined as brain damage occurred within 72 hours after SAH) plays a key role in symptom development following SAH (Chen et al., 2018, 2019).The probable underlying mechanisms for EBI include apoptosis, necroptosis, direct neuronal death, and autophagy (Chen et al., 2016a, 2019; Zhou et al., 2021).Nevertheless, Zille et al.(2017) found that inhibitors of parthanatos, mitophagy, protein or mRNA synthesis, autophagy,or caspase-dependent apoptosis did not affect intracerebral hemorrhagein vivoorin vitro.Thus, the extent to which various kinds of cell death lead to SAH-induced toxicity remains unclear.
Obstructive sleep apnea (OSA) can induce the formation of intracranial aneurysms, promote aneurysm rupture, aggravate EBI, and worsen the general outcome of patients with vascular aneurysms after SAH (Mason et al., 2011; Chernyshev et al., 2019; Zaremba et al., 2019).In a retrospective analysis, Schuiling et al.(2005) reported that 34% of SAH patients exhibited OSA symptoms, including snoring and more frequent nocturnal awakenings.Bir et al.(2018) also reported that most complications that occur after rupture of an intracranial aneurysm are the consequence of uncontrolledOSA, and that SAH combined with OSA can aggravate brain injury and lead to a worse outcome.However, the mechanism by which OSA aggravates brain injury remains unclear.
It is unknown whether SAH or OSA leads to hypoxic-ischemic brain injury(Xu et al., 2016; Yu et al., 2019).Xiong et al.(2021) reported that OSA increased oxidative stress and inflammation, then worsened bleomycininduced pulmonary fibrosis.Hypoxia-inducible factor-1α (HIF-1α) is an important transcription factor whose expression is upregulated in mammals and humans in response to hypoxia or ischemia.Activated HIF-1α enters the nucleus, where it regulates target gene transcription (Xu et al., 2016; Yu et al., 2019).Increasing evidence demonstrates that HIF-1α is significantly upregulated after SAH, and that inhibiting HIF-1α expression can alleviate EBI (Li et al., 2014; Zhaba et al., 2021).In addition, HIF-1α is significantly upregulated in patients with OSA, and hypoxia/ischemia-induced injury is alleviated by HIF-1α inhibition (Yu et al., 2019; Gabryelska et al., 2020).Yu et al.(2019) also reported that hypoxia can induce reactive oxygen species(ROS) overproduction and accumulation, which contribute to cell pyroptosis by activating the HIF-1α and nuclear transcription factor-κB (NF-κB) signaling pathways.
Pyroptosis is a recently recognized mechanism of programmed cell death that has been comprehensively studied which plays a key role in neurological damage (Fricker et al., 2018).Apoptosis-related inhibitors cannot prevent pyroptosis without causing calcium ion overload, which play a vital important role in the EBI after SAH (Chen et al., 2021).In recent years, pyroptosis has been reported to be involved in cerebral hemorrhage, tumors of the central nervous system, and traumatic brain injury (Gao et al., 2020; Lien et al.,2021; Xu et al., 2021b).Yu et al.(2019) demonstrated that pyroptosis is a very important cell death mechanism in hypoxia-induced tissue injury and cell death, particularly in hypoxia-related diseases and OSA.Nonetheless,the exact function of pyroptosis in SAH, and whether it is a viable target for therapy, remains to be verified.Furthermore, a comprehensive investigation of the relationship between OSA on SAH is lacking, and the specific mechanism underlying this relationship is unclear (Mason et al., 2011;Chernyshev et al., 2019; Zaremba et al., 2019).In this study, we explored the function of OSA in EBI following experimentally-induced SAH in mice.
Materials and Methods
Animals
All of the animal experiments carried out as part of this research project adhered to the guidelines stipulated by the National Institutes of Health regarding the care of laboratory animals.Approval for the animal experiments was obtained from the Ethics Committee of the Nanjing Medical University,China on January 15, 2020 (approval No.NYLL-2020-11).
A previous study reported that estrogen levels affect EBI after SAH (Kao et al., 2013).Thus, the animals used for this study were 120 healthy adult male C57BL/6J mice (age 6-8 weeks; weight 22-25 g, Nanjing Medical University,China, license No.SCXK (Su) 2019-00001).The mice were housed in a climatecontrolled environment at 25 ± 2°C and 55 ± 5% humidity with 12/12-hour dark/light cycle and unrestricted access to water and food.
Experimental subarachnoid hemorrhage model
The 120 mice were randomly divided into eight groups (15 animals/group):sham, OSA, SAH, SAH + OSA, SAH + si-Con, SAH + si-HIF-1α, SAH + OSA + si-Con, and SAH + OSA + si-HIF-1α.The SAH model was established by the endovascular perforation method, based on a procedure that has been previously described (Chen et al., 2021).To anesthetize the male C57BL6/J mice, pentobarbital sodium (Sigma, St.Louis, MO, USA) was administered via intraperitoneal injection at a dose of 50 mg/kg.The anesthetized animals were placed on a heating pad during the operation to maintain their temperature at 37 ± 0.5°C.A midline incision was made in the neck to expose the left internal and external carotid arteries.The left external carotid artery was then ligated and cut, resulting in a 3-mm stump.A 15-mm-long 4-0 monofilament nylon suture was introduced into the left internal carotid artery via the external carotid artery stump to perforate the cerebral artery at the midpoint and where it bifurcated from the anterior artery.Then, the suture was inserted another 3 mm to induce further perforation at these two sites.The suture was removed after approximately 10 seconds.The sham mice underwent the same surgical procedure without perforation.
Experimental OSA model
After the mice recovered from the SAH surgery, OSA was induced as we described in a previous study (Yu et al., 2019).Briefly, the mice were exposed to 8 hours of intermittent normoxic or hypoxic air conditions each day from the day after the SAH procedure.For the OSA mice, the concentration of oxygen in the mouse compartments was monitored utilizing an O2analyzer(Meicheng, Shanghai, China, CY-12C).Manipulation of reoxygenation and hypoxia during the daytime was achieved by varying the concentrations of nitrogen and oxygen.The cycles (30 cycles/hour) of intermittent hypoxia entailed 2 minutes of hypoxia at 7 ± 1% O2followed by reoxygenation at 21 ±0.5% O2.The control mice were subjected to normoxic air conditions.After 48 hours of atmospheric manipulation, the mice were euthanized.
Assessment of mortality and SAH grade
We evaluated the mortality of the mice 48 hours after SAH/OSA.For the surviving mice, the SAH grade was determined based on a grading system described previously (Sugawara et al., 2008).The scores ranged from 0 to 18,with a higher scores indicating better neurological function.Mice with SAH grading scores below 7 and those with no obvious brain injury were excluded from the following experiments (Sugawara et al., 2008; Chen et al., 2021).
Neurological function assessment
The seriousness of EBI was assessed at 48 hours following SAH utilizing a neurological grading system that we described previously (Chen et al., 2020a).The scoring system comprised six tests and explicit protocols, and is illustrated in Additional Table 1.Neurological function scores ranged from 3 to 18.Behavioral assessment was performed for all of the mice in the two cohorts,with higher scores representing better neurological function.
Brain water content measurement
As brain edema is an important factor leading to EBI, brain water content was evaluated utilizing the standard wet-dry technique, as described previously(Chen et al., 2019, 2020a, 2021).The mice were euthanized with 100 mg/kg pentobarbital sodium via intraperitoneal injection 48 hours after SAH/OSA,and whole brains were collected, weighed (to obtain the wet weight) and separated into the brain stem, cerebellum, right cortex, and left cortex.These samples were then dried in an oven (Shanghai Bluepard Instruments Company, Shanghai, China) at 100°C for 24 hours and weighed (to obtain the dry weight).The percentage of brain water content was calculated as follows:(wet weight - dry weight)/wet weight × 100.
TdT-mediated dUTP-biotin nick end labeling assay
A TdT-mediated dUTP-biotin nick end labeling (TUNEL) assay was carried out to evaluate cell death in the brain cortex.The procedure was performed with a TUNEL staining kit according to the manufacturer’s instructions (Roche Diagnostics GmbH, Basel, Switzerland; Cat# 1684817).Briefly, 50 µL of TUNEL reaction mixture was added to each slide, which were then incubated in a humidified dark chamber at 37°C for 60 minutes.Next, the slides were incubated with 4′,6-diamidino-2-phenylindole at ambient temperature for 5 minutes in the dark to stain the nuclei.A fluorescence microscope (Carl Zeiss,Jena, Thuringia, Germany) was utilized for imaging.The apoptotic index (%)was calculated as follows: number of TUNEL-positive cells/total number of cells × 100.
Cytokine measurements
48 hours after SAH/OSA, interleukin (IL)-1β (Abcam, Cambridge, MA,USA, Cat# ab197742), IL-18 (Abcam, Cat# ab216165), IL-6 (Abcam, Cat#ab222503), and NF-κB (Abcam, Cat# ab176663) levels in the cerebral cortex were evaluated by enzyme-linked immunosorbent assay according to the manufacturer’s instructions.
Mouse treatment and cell transfection with small interfering RNA
Pentobarbital sodium was used to anesthetize the mice, which were then restrained on a stereotaxic apparatus (Narishige, Tokyo, Japan).Subsequently, a burr hole was created in the left hemisphere utilizing the following coordinates: 1 mm lateral, 0.2 mm posterior, and 2.2 mm below the horizontal plane of the bregma.This was followed by the injection of 5 µL of small interfering RNA (siRNA) into the left lateral ventricle at a rate of 0.5 µL per minute.To promote the silencing effect, the siRNA was injected 48 hours before SAH.Cell transfection was performed utilizing Lipofectamine RNAiMax reagent (Thermo Fisher Scientific, Waltham, MA, USA) in Opti-MEM medium according to the manufacturer’s instructions.The sequences of the targeted siRNA and control siRNAs, which were manufactured by JiKai (Shanghai,China), were as follows: si-HIF-1α sense, 5′-GCU GUU CAC UAA AGU GGA AUC-3′; negative control siRNA sense, 5′-UUC UCC GAA CGU GUC ACG UTT-3′.
Cultured cell lines
All cell lines were cultured as described previously (Chen et al., 2021).HT-22 cells (RRID:CVCL_0321) were provided by Anhui Medical University (Wuxi,China).HT-22 cells were cultured at 37°C with a CO2concentration of 5%in Dulbecco’s modified Eagle’s medium, 10% fetal bovine serum (Gibco,Thermo Fisher Scientific), and 1% penicillin-streptomycin.These experiments were carried out at a cell density ranging between 60% and 80%.The HT-22 cells were authenticated by sensitivity to hemin-induced damage and visually assessed morphology (Alim et al., 2019).All cell lines used in these experiments had been passaged less than 25 times.
HT-22 SAH model
For the HT-22 SAH experiments, hemin (20 to 140 mM for 48 hours; Cat#H9039, Sigma) was used to induce HT-22 cell death.The hemin was dissolved in 0.1 M NaOH.Cells grown to 60% to 70% confluency were treated with 80 mM hemin (in NaOH, median lethal dose, LD50) dissolved in ddH2O.Dimethyl sulfoxide was added to the hemin solution at a final concentration of 80 mM, and the combined solution was sterilized by passing it through a 0.22-µm filter before applying it to the cells.After 48 hours of treatment, the HT-22 cells were washed with phosphate-buffered saline (PBS) pre-warmed to 37°C and evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay and dead/live assay to assess cell viability, according to the assay manufacturers’ instructions (Promega, Madison, WI, USA).As reported previously, treating neurons with 80 mM hemin for 48 hours imitates SAH or intracranial hemorrhage conditions (Alim et al., 2019).
MTT assay
Cell viability was assessed by MTT assay.HT-22 cells were seeded into 96-well plates, followed by the addition of 20 µL MTT (Sigma) to each well and incubation at 37°C for 4 hours.Then, the culture supernatant was removed,and the formazan crystals were lysed with dimethylsulfoxide (Sigma) by agitating the plate gently.Afterward, the absorbance at 490 nm was evaluated using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA), and the results were normalized to the vehicle group.
Dead/live assay
In the case of the dead/live assay, HT-22 cells were washed twice followed by resuspension in PBS.The cells were then treated with calcein-AM (3µM) and propidium iodide (PI, 4 µM) for 20 minutes in the dark at ambient temperature.Next, the cells were washed and resuspended in PBS and analyzed using a fluorescence microscope system (DMIL 4000B, Leica,Weitzlar, Hesse, Germany).Each experiment was performed in triplicate.
Real-time polymerase chain reaction
Apoptosis-associated speck-like protein containing a CARD (ASC) is an adapter protein encoded by the PYCARD gene that facilitates the assembly of several inflammasomes; first, it forms a caspase-1-activating scaffold,which is followed by the cleavage of pro-caspase-1 to active caspase-1, which ultimately leads to activation of IL-1β and IL-18 and induction of pyroptosis(de Almeida et al., 2015; Liang et al., 2020).HIF-1α plays an important role in neuroinflammation after OSA and SAH (Ostrowski et al., 2005; Xiong et al., 2021).Real-time PCR was used to detect the expression of HIF-1α and ASC.Total RNA was extracted from the fresh left cortex samples obtained 48 hours after SAH/OSA from the cortex samples or from HT-22 cells, using TRIzol reagent according to the manufacturer’s instructions.Quantitative real-time polymerase chain reaction (PCR) was performed using standard procedures, as described previously (Chen et al., 2021).Subsequently, reverse transcription of RNA to complementary DNA was performed using a reverse transcription kit (Takara, Kyoto, Japan).mRNA levels were determined by qPCR utilizing SYBR Green Master Mix (Toyobo Co., Ltd., Osaka, Japan).mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).All analyses were performed in triplicate.The sequences of the primers used to amplify HIF-1α and ASC were as follows: HIF-1α, forward, 5′-GGA CAA GTC ACC ACA GGA CA-3′ and reverse, 5′-GGG AGA AAA TCA AGT CGT GC-3′; ASC,forward, 5′-GGC ACA GCC AGA ACA GAA CA-3′ and reverse, 5′-GCA CGA ACT GCC TGG TAC TG-3′; GAPDH, forward, 5′-TGA TTC TAC CCA CGG CAA GTT-3′ and reverse, 5′-TGA TGG GTT TCC CAT TGA TGA-3′.The 2-∆∆Ctmethod was used to assess the RNA expression levels (Livak and Schmittgen, 2001).
Lactate dehydrogenase release
In vitroneurotoxicity was ascertained by evaluating the release of lactate dehydrogenase (LDH) using a kit according to the manufacturer’s instructions(Jiancheng bioengineer Institute, Nanjing, China).HT-22 cells were collected immediately after SAH/OSA exposure, followed by incubation with the LDH release reagent (Beyotime, Shanghai, China) for 1 hour.After centrifugation at 1000 ×g, the cell supernatant was combined with an LDH detection reagent (Beyotime) and incubated at room temperature in the dark for 30 minutes.The LDH concentration was quantified (Multiskan MK3, Thermo Fisher Scientific) by measuring the absorbance at 490 nm according to the manufacturer’s instructions (Chen et al., 2020b), and the results were normalized to the control group.
Immunohistochemical analysis of glial fibrillary acidic protein
Paraffin-embedded slices of the cerebral cortex were cut into 5-µm-thick sections.After treatment with blocking serum, the sections were incubated with rabbit polyclonal anti-glial fibrillary acidic protein (GFAP; 1:30, Abcam,Cat# ab7260, RRID: AB_305808) antibody overnight at 4°C.After washing with PBS containing Tween-20 three times, the samples were incubated with the secondary antibody at 37°C for 1 hour.Then, 4′,6-diamidino-2-phenylindole(10 µg/mL) was used to stain the nuclei, and images were obtained using a Zeiss fluorescence imaging microscope (Carl Zeiss, Jena, Thuringia, Germany).
Western blot assay
Western blotting was performed to detect pyroptosis-associated proteins, as described previously (Chen et al., 2016a, 2018, 2019, 2020a).Total protein was extracted from HT-22 cells or fresh left cortex samples.The protein concentrations were evaluated by the BCA method (Invitrogen, Waltham,MA, USA).Subsequently, the proteins (50 µg) were resolved on a sodium dodecyl sulfate-polyacrylamide gel and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), followed by blocking with 5% nonfat milk in TBS-T (Tween 20, 0.1%) at 20°C for 1 hour.The membranes were incubated overnight at 4°C with the primary antibodies: rabbit anti-HIF-1α(1:1000, monoclonal, Abcam, Cat# ab179483, RRID: AB_2732807), rabbit anti-pro-caspase-1 p20 (1:1000, Abcam, Cat# ab238972, RRID: AB_2884954),rabbit anti-β-actin (1:1000, polyclonal, Abcam, Cat# ab8227, RRID:AB_2305186), rabbit anti-nucleotide-binding domain leucine-rich repeat pyrin domain containing 3 (NLRP3; 1:1000, Abcam, Cat# ab263899, RRID:AB_2889890), rabbit anti-caspase-1 (1:1000, Abcam, Cat# ab108362, RRID:AB_10858984), and rabbit anti-ASC (1:1000, Cell Signaling Technology, Cat#67824, RRID: AB_2799736).The membranes were washed with TBS-T three times, and then incubated with goat anti-mouse IgG secondary antibodies(1:5000, Abcam, Cat# ab97035, RRID: AB_10680176) for 90 minutes at 20°C.Quantitative image analysis was performed using ImageJ version 1.52(Schneider et al., 2012).
Statistical analysis
No statistical methods were used to predetermine sample sizes; however,our sample sizes are similar to those reported in previous publications (Chen et al., 2019, 2021).The evaluator was blinded to the grouping.Nine mice died after the SAH procedure; the rest of the mice were randomly assigned to the groups at up to 15 mice per group.The data are reported as the mean ± standard error of the mean (SEM).SPSS 14.0 (IBM, Armonk, NY,USA) and GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA, www graphpad.com) were used for the statistical analyses.Differences between multiple groups were analyzed using one-way analysis of variance followed by Tukey’spost hoctest.For all statistical analyses, differences were considered significant atP< 0.05.
Results
Mortality and SAH grade after SAH or OSA
Earlier clinical research had reported that OSA can aggravate EBI, increase mortality, and worsen overall outcome in SAH patients (Mason et al.,2011; Chernyshev et al., 2019; Zaremba et al., 2019).Thus, we used the endovascular perforation technique followed by atmospheric manipulation to induce SAH and OSA in mice (Figure 1A).We then assessed the influence of OSA on parameters such as SAH grade and survival.As illustrated in Figure 1, survival rates were lower in the SAH + OSA cohort, although there was no significant difference compared with the SAH cohort (OR = 0.5, 95% CI:0.095-2.628, Figure 1B).There was no significant difference in SAH grades between the SAH and SAH + OSA groups (P> 0.05; Figure 1C).Even though OSA did not increase SAH grade or decrease survival, there was a clear trend toward aggravation of both parameters, suggesting a possible correlation that may not have reached statistical significance because of the small sample size.
OSA aggravates EBI following SAH in vivo
To elucidate the effects of OSA after SAH on EBI, we measured brain water content 48 hours after SAH to assess brain damage.Compared with the control group, SAH considerably increased the brain water content, and this effect was aggravated by OSA (Figure 2A).The neurological scores were lower in the SAH group compared with the sham group, and the SAH + OSA exhibited significantly worse neurological scores compared with the SAH group (P< 0.05; Figure 2B).To detect hippocampus damage following SAH and OSA, we used the TUNEL assay to quantify cell death 48 hours after injury.As anticipated, there was more hippocampal cell death in the SAH+ OSA group than in the SAH group (Figure 2C).Taken together, these data show that OSA aggravates EBI after SAH.
OSA aggravates hemin-induced HT-22 cell damage
Given that OSA aggravates EBIin vivo, we next sought to elucidate the neuronal damage caused by OSAin vitro.Hemin was used to induce SAH,and a cell OSA technique was used to induce OSA (Figure 1A).The MTT assay results showed that the rate of neuronal death increased significantly after treatment with hemin, and that this effect was aggravated by OSA (Figure 3A).SAH increased LDH release, and OSA enhanced this effect (Figure 3B).Furthermore, the results from the dead/live assay illustrated that OSA aggravated hemin-induced HT-22 cell damage (Figure 3C).
OSA aggravates neuroinflammation after SAH in vivo
Earlier studies have demonstrated that neuroinflammation is a critical feature of EBI following SAH, and that increased neuroinflammation can aggravate EBI(Wei et al., 2020; Chen et al., 2021).The inflammatory complex can induce the release of pro-inflammatory cytokines and/or the initiation of pyroptosis(Chen et al., 2021).Thus, we assessed the hippocampal levels of IL-1β, IL-18,IL-6, and NF-κB by enzyme-linked immunosorbent assay.The results showed that IL-1β, IL-18, IL-6, and NF-κB levels were increased considerably in the SAH group compared with the sham group, whereas levels of pro-inflammatory cytokines were further increased in the OSA + SAH group compared with those in the SAH group (P< 0.05; Figure 4A-D).Immunohistochemical analysis also illustrated that GFAP expression in the cerebral cortex and astrocyte proliferation increased after SAH, and that OSA aggravated these effects (Figure 4E).This suggests that OSA may be aggravated by the elevated levels of pro-inflammatory cytokines, and, at the same, time, OSA may delay clearance of pro-inflammatory cytokines.
OSA aggravates pyroptosis after SAH in vivo
Previous studies have indicated that pyroptosis is an important form of cell death in diseases of the central nervous system, and that it also plays an important role in SAH and OSA (Yu et al., 2019; Chen et al., 2021; Xu et al.,2021a).Thus, we next examined the expression of the pyroptosis-associated proteins caspase-1, pro-caspase-1, and NLRP3 following SAH and OSA in fresh left cortex samples by western blot assay (Figure 5A).Compared with the sham and OSA groups, caspase-1, pro-caspase-1, and NLRP3 levels were increased in the SAH group (P< 0.05; Figure 5B-D) and were even higher in the SAH + OSA group (P< 0.05; Figure 5B-D).
OSA aggravates pyroptosis following SAH via the HIF-1α/ASC signaling pathway in vivo
As HIF-1α is vital to both OSA and SAH (Xu et al., 2016; Yu et al., 2019), we hypothesized that activation of the HIF-1α signaling pathway was the main reason for aggravation of pyroptosis by OSA in mice that have experienced SAH.Wu et al.(2020) reported that ASC can modulate HIF-1α stability, thereby modifying the expression levels of NLRP3 inflammasome components.To clarify the potential mechanism of pyroptosis and the role of the HIF-1α/ASC signaling pathway following SAH/OSA, we detected HIF-1α and ASC gene andprotein expression levels in fresh left cortex by real-time PCR and western blot assay, respectively.The results showed that HIF-1α expression was elevated after 48 hours in the SAH group, and was significantly higher in the OSA +SAH group compared with the SAH group (P< 0.05; Figure 6A and B).ASC and HIF-1α protein expression levels were detected by western blot (Figure 6C), which showed that both were increased significantly in the OSA + SAH group compared with the SAH group (P< 0.05; Figure 6D and E).Hence, OSA promotes EBI after SAH by aggravating pyroptosis, possibly through the HIF-1α/ASC signaling pathway.
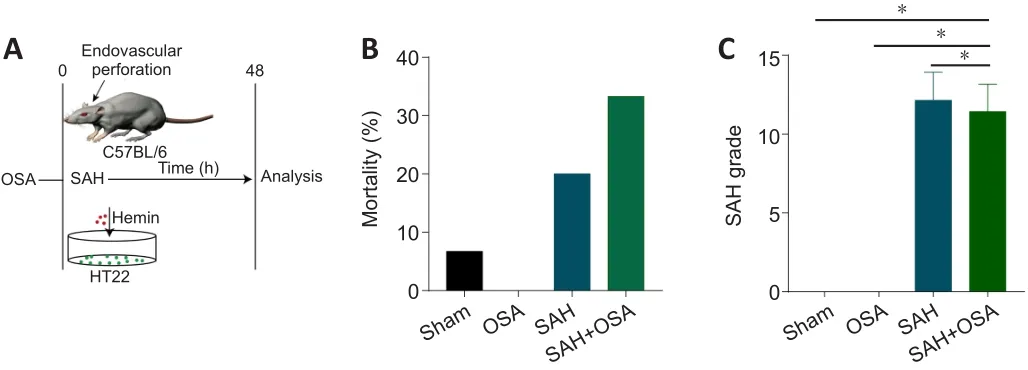
Figure 1|Mortality and SAH grade after SAH and OSA.
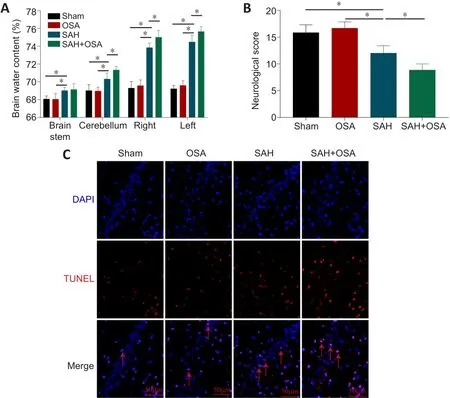
Figure 2|OSA aggravates EBI following SAH in vivo.
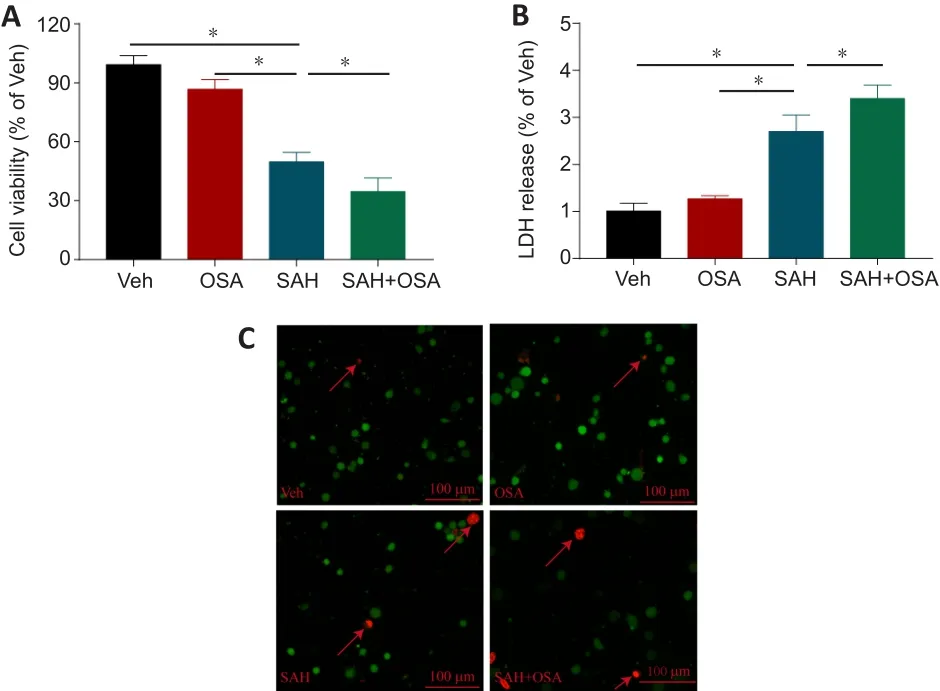
Figure 3|OSA increases hemin-induced HT-22 neuronal cell injury in vitro.
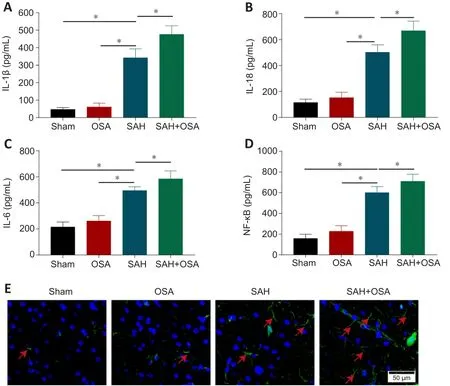
Figure 4| OSA aggravates neuroinflammation after SAH in vivo.
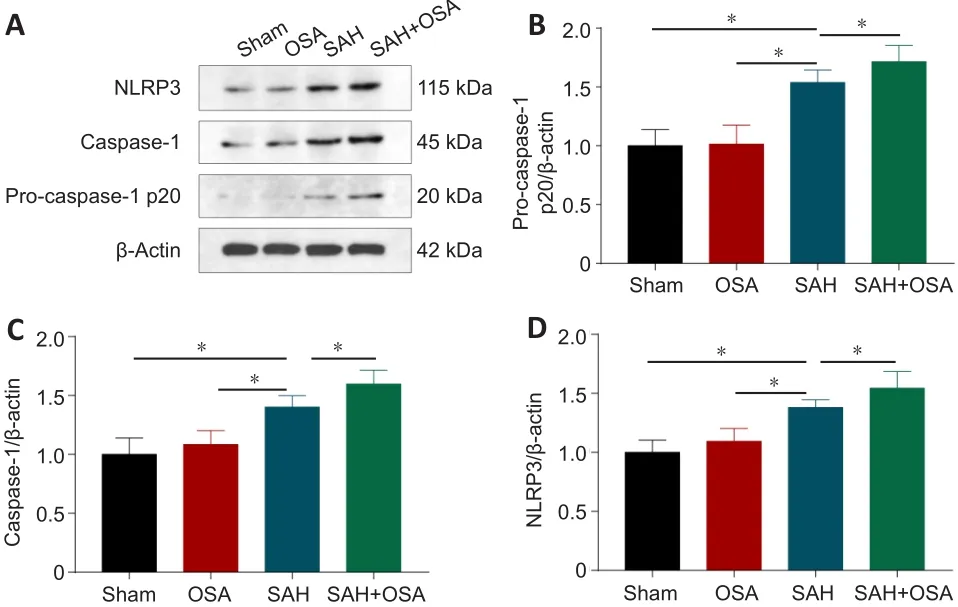
Figure 5| OSA aggravates pyroptosis after SAH in vivo.
Knockdown of HIF-1α alleviates pyroptosis after SAH and OSA in vitro
To ascertain the effect of HIF-1α on pyroptosis following SAH/OSA, the expression of pyroptosis-associated proteins was assessed by western blot(Figure 7A).Compare with the si-Con group, HIF-1α knockdown considerably reduced the expression of caspase-1, pro-caspase-1, and NLRP3 in the SAH+ OSA group (P< 0.05; Figure 7B-D).The MTT assay results showed that neuronal death was significantly decreased in the si-HIF-1α group compared with the si-Con group (P< 0.05; Figure 7E).Furthermore, LDH release was significantly decreased in the si-HIF-1α group compared with the si-Con group (P< 0.05; Figure 7F).The dead/live assay results illustrated that hemininduced HT-22 cell damage was significantly decreased in the si-HIF-1α group compared with the si-Con group (P< 0.05; Figure 7G).
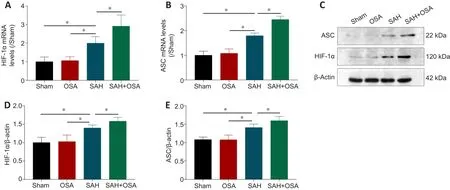
Figure 6|OSA aggravates pyroptosis after SAH in vivo via the HIF-1α/ASC signaling pathway.
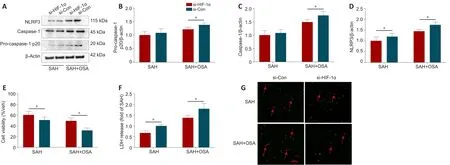
Figure 7| HIF-1α knockdown alleviates pyroptosis after SAH/OSA in vitro.
Knockdown of HIF-1α alleviates EBI after SAH and OSA
To further elucidate the effects of HIF-1α knockdown on EBI in mice that have undergone SAH/OSA, we assessed neurological score, TUNEL assay results,and brain water content at 48 hours following SAH.The results showed that HIF-1α knockdown considerably increased the neurological score compared with that in the si-Con group (P< 0.05; Figure 8A).The brain water content was decreased in the HIF-1α knockdown group compared with the si-Con group (P< 0.05; Figure 8B).To further assess hippocampal neuron death following HIF-1α knockdown, we performed a TUNEL assay to quantify cell death.As anticipated, less hippocampal tissue damage occurred in the si-HIF-1α group compared with in the si-Con group (Figure 8C).These findings suggest that HIF-1α knockdown can alleviate EBI after SAH/OSAin vivo.
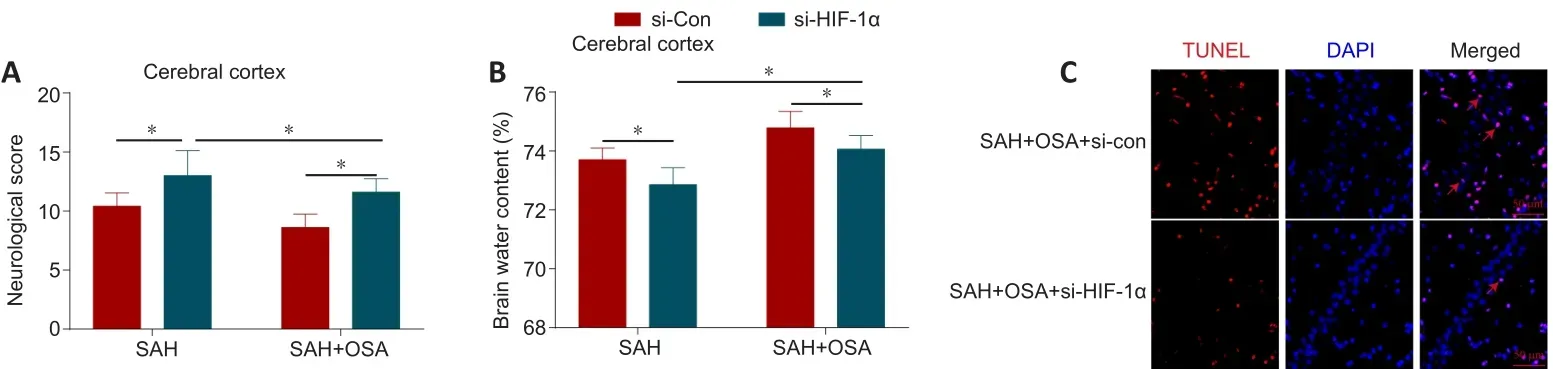
Figure 8| HIF-1α knockdown alleviates EBI after SAH and OSA in vivo.
Discussion
In this study, we found that OSA aggravates EBI in a mouse model of SAH.Specifically: (1) OSA aggravates neurological dysfunction after SAH; (2) OSA enhances brain damage after SAH; (3) OSA promotes neuroinflammation after SAH, thereby increasing inflammatory damage to the brain; (4) OSA can activate pyroptosis after SAH and induce neuronal death; and (5) the mechanism by which OSA aggravates EBI after SAH may be related to the HIF-1α/ASC signaling pathway.
Interruption of breathing by OSA causes intermittent hypoxia, resulting in decreased blood oxygen saturation and impaired sleep quality, and prolonged hypoxia may induce various inflammatory responses that affect the function of the vascular endothelium (Tan et al., 2021).OSA seriously impairs the quality of life of older people and also leads to or aggravates various comorbidities, including hypertension (Prabhakar et al., 2020), coronary heart disease (Strausz et al., 2021), ICH (Orrù et al., 2020), and stroke (Li et al.,2020).The molecular mechanism underlying OSA-induced disease has proven extremely complicated to unravel, and there are several theories that attemptto explain this process.OSA can induce activation of the inflammatory system,leading to the production of high levels of ROS, and ROS are a probable therapeutic target for alleviating hypoxia-induced tissue injury and cell death(Yu et al., 2019).Furthermore, increasing evidence confirms the relationships between OSA, cognitive impairment, and central nervous system disease(Hung et al., 2008; Pontes-Neto et al., 2010; Mason et al., 2011; Chernyshev et al., 2019; Zaremba et al., 2019; Orrù et al., 2020; Geer et al., 2021).
Chen et al.(2021) reported that inflammasome activation and pyroptosis are largely responsible for the neuronal death and EBI observed in the experimental mouse model of SAH.Inin vivoandin vitromodels of SAH,the expression of triggering receptor on myeloid cells can trigger microglial pyroptosis by activating the NLRP3 inflammasome and aggravating neuroinflammation (Xu et al., 2021a).Polarization of microglia is the primary factor that leads to neuroinflammatory damage following stroke.The NLRP1 and NLRP3 inflammasomes, which are members of the NLR inflammasome family, are expressed mainly within the microglia, neurons, and endothelium,and play crucial roles in diseases of the central nervous system (Tan et al.,2014; Lin et al., 2018; Fang et al., 2020).Yuan et al.(2020) also reported that gasdermin D can induce pyroptosis after SAH, that the absent in melanoma 2(AIM2) inflammasome and AIM2/caspase-1 pathway mediated EBI following SAH, and that the degree of EBI was lessened when AIM2 and caspase-1 were knocked down.In the current study, we found that OSA can activate pyroptosis after SAH and induce neuronal death, and that these effects may be mediated by the HIF-1α/ASC signaling pathway.A previous study also reported that hypoxia or OSA can induce ROS production, which then contributes to pyroptosis via the NF-κB/HIF-1α signaling pathway (Yu et al.,2019).
Similarly, neuroinflammation functions synergistically with pyroptosis in the pathogenesis of OSA/SAH-induced EBI.In a murine model of sleep apnea, the pyroptosis-associated NLRP3 inflammasome was shown to regulate PINK1-Parkin-depended mitophagy, and NLRP3 knockout or inhibition improved neurocognitive function after OSA through alleviating neuroinflammation and enhancing Parkin-mediated mitophagy (Wu et al., 2021).Gong et al.(2020)also reported that OSA causes IH in mice, then leads to neuronal apoptosis and hippocampal neuroinflammation, affecting spatial learning and memory;these effects were, reversed by pinocembrin-mediated inhibition of the NLRP3 inflammasome and promotion of BNIP3 (Bcl-2/adenovirus E1B 19-kDa interacting protein 3)-mediated mitophagy in the hippocampus.Moreover, in an experimental model of diabetes combined with OSA, neuroinflammation and hippocampal neuronal apoptosis were the main reasons for the cognitive deficits that were observed, and these effects were thought to be mediated through the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase-3β signaling pathway (Shi et al., 2018).Thus, our findings suggest that OSA can aggravate SAH-induced EBI by activating neuroinflammation and pyroptosis.
Although many studies have shown that pyroptosis and neuroinflammation are involved in a variety of diseases of the central nervous system, the mechanisms and molecular networks regulating the interactions between these two processes remained unclear.In this study, we demonstrated that pyroptosis and neuroinflammation lead to neuronal death after SAH/OSA, that neuronal death is aggravated by OSA, and that the mechanism is potentially dependent on the HIF-1α/ASC signaling pathway.Wu et al.(2020)also reported that ASC can modulate HIF-1α stability, causing lymph node metastasis of oral squamous cell carcinoma.A previous study had confirmed that downregulation of the expression of HIF-1α and its target genes can decrease neuronal apoptosis and BBB disruption in an animal model of SAH (Xu et al., 2016).In a mouse model of ischemic stroke, neuron-specific knockout of HIF-1α decreases early acute neurological impairment and neuronal cell death by decreasing apoptosis and increasing angiogenesis (Barteczek et al.,2017).Additionally, multiple previous studies have demonstrated that many neuroprotective drugs can inhibit HIF-1α expression, thereby improving neurological dysfunction, decreasing neuronal death, and reducing bloodbrain barrier permeability (Schaible et al., 2014; Trollmann et al., 2014; Min et al., 2015).Hu et al.(2021) reported that a particular synthetic TGR5 agonist,INT-777, can alleviate NLRP3-ASC inflammasome activation and reduce neuroinflammation following SAH.Ystgaard et al.(2019) reported that brain infarction volumes decreased significantly after ASC was knocked out in a neonatal hypoxia-ischemia model.Hence, the HIF-1α/ASC signaling pathway has a crucial function in neuroinflammation and pyroptosis after SAH.
The present study has several limitations.First, we did not determine the precise mechanism of the ASC/HIF-1α axis in regulating pyroptosis.Second,we did not explore the detailed basis of neuroinflammation, such as activation of microglial cells or astrocytes.Finally, it is difficult to accurately evaluate microglial activation using immunofluorescence alone, and additional methods such as flow cytometry may be more suitable and accurate for use in future investigations.
In summary, this study showed that OSA can worsen neurological outcomes,increase brain edema, aggravate neuronal damage, and promote neuronal death in mice that have undergone SAH.These effects are potentially mediated by enhanced neural pyroptosis and neuroinflammation, specifically through the action of ASC and the NLRP3 inflammasome, which is emerging as a crucial cellular regulatory mechanism that contributes to EBI following SAH.This study also showed that OSA aggravates neuroinflammation,pyroptosis, and neuronal death through the HIF-1α/ASC pathway.
Acknowledgments:We thank Junhui Chen (Anhui Medical University, China)for providing the HT-22 cells.
Author contributions:Study design and manuscript revision: LY, JX, CFZ;experiment implementation: LY, JX, CFZ, QL, SM, JJJ, YS, QL, WG; manuscript draft: LY, JX, CFZ; data analysis: LY, JX, QL, SM, YS, QL, WG.All authors read and approved the final manuscript.
Conflicts of interest:The authors declare that they have no competing interests.
Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Junfan Chen, The Chinese University of Hong Kong,China; Clarissa Cavarsan, Universidade Federal do Paraná, Brazil.
Additional files:
Additional Table 1:Neurological Behavior Scores.
Additional file 1:Open peer review reports 1 and 2.
- 中国神经再生研究(英文版)的其它文章
- Modeling Alzheimer’s disease:considerations for a better translational and replicable mouse model
- Engineering cerebral folding in brain organoids
- Verapamil, a possible repurposed therapeutic candidate for stroke under hyperglycemia
- Delayed activation of leg somatotopic fibers of an injured corticospinal tract in a patient with cerebral infarction
- Polydopamine-modified chitin conduits with sustained release of bioactive peptides enhance peripheral nerve regeneration in rats
- NOVA1 promotes SMN2 exon 7 splicing by binding the UCAC motif and increases SMN protein expression