
基于TF-mRNA调控网络识别消化道癌症共享功能模块*
2022-03-16 13:00蓝树金饶绍奇
肿瘤预防与治疗 2022年2期
蓝树金,饶绍奇
523808广东 东莞,广东医科大学 公共卫生学院
消化道癌症包含食管癌、胃癌、结肠癌和直肠癌,这几种消化道癌症对人体健康造成巨大威胁,这几种癌症病因及发病机制复杂[1]。国内学者对上消化道癌症的研究指出中国上消化道恶性肿瘤新发病例占全球的44.60%,这一数据表明消化道癌癌症对医疗卫生系统的及人民健康造成巨大的负担[2-3]。但受限于当前医疗水平的发展,对于这些癌症的常规治疗方案主要是外科手术切除和化疗,但是癌症的高复发率会影响常规治疗方案的最终效果并有可能造成手术失败,影响医疗效果。随着测序技术的快速发展,利用生物信息学可以分析疾病的分子机制及识别重要的分子机制以及重要分子功能。
癌症患者的预后往往是由疾病的分型决定的,而肿瘤的最终的分型是由多种基因调节网络以及不同组学之间的相互影响最终决定的[4]。转录调控因子(transcription factor,TF),亦称为反式作用因子,是有序结合在目标基因启动子序列中的特殊位点,其通过结合基因的特定序列motif,亦称作模序,在转录前水平调节下游基因的表达水平[5-6]。已有大量相关研究指出TF在转录调控网络中起重要作用,如Farhan等[7]发现FOXO转录因子在癌症代谢及血管生成中起作用,Lan等[8]发现CIN-like TCP作为植物特定的转录因子在植物的形态发育起重要作用,针对植物形态诸如植物枝叶的发育等对调控信息。所列举的研究均表明了转录因子在细胞乃至形态学上都起到重要的作用。通过构建疾病相关的TF-mRNA调控网络,识别在调控网络中起重要作用的TF及其靶基因,并探究TF对其调控的靶基因的作用参与的生物学过程以及疾病相关的致病机制有助进一步在疾病发生发展转录前水平转录调控模式。有研究指出转录因子在癌症的成因、发展以及恶化都起了重要作用,Zacksenhaus等[5]的研究指出在乳腺癌中,影响调节因子或染色体形态关键基因的变异在基因的增强子和促进子中形成了新的TF结合位点,并间接促进了癌症的发生。
消化道癌症包括食管癌、胃癌、结肠癌以及直肠癌,这几种癌症具有一些共同的背景特点,首先在组织胚胎学上,消化道如食管,胃及结直肠具有相似的生长发育背景,均起源于内胚层,其次,尽管食管、胃、结肠及直肠在消化功能各异,但在解剖结构上,其解剖结构大体都是管状结构[9]。通过系统分析消化道癌症的共享分子并构建TF-mRNA调节网络可分析出重要的分子以及重要的功能。
本研究旨在通过TF-mRNA调控网络系统分析消化道癌症,即包含食管癌、胃癌、结肠癌及直肠癌之间共享的转录调控模式及其所调控基因的参与的生物学功能,结合转录调节网络及蛋白互作网络,识别重要消化道癌症共享转录因子及其靶基因并为消化道癌症治疗方法提供研究方向。
1 材料与步骤
1.1 数据获取
消化道癌症(食管癌、胃癌、结肠癌及直肠癌)RNAseq数据都是由TCGA公共数据网站下载。分别选取了STAD,ESCA, COAD及READ四种癌症项目,其分别对应胃癌、食管癌、结肠癌及直肠癌,本研究下载了对应每种癌症的实验策略为RNAseq,工作类型为HTSeq-Counts数据,癌症基本信息如表1所示。
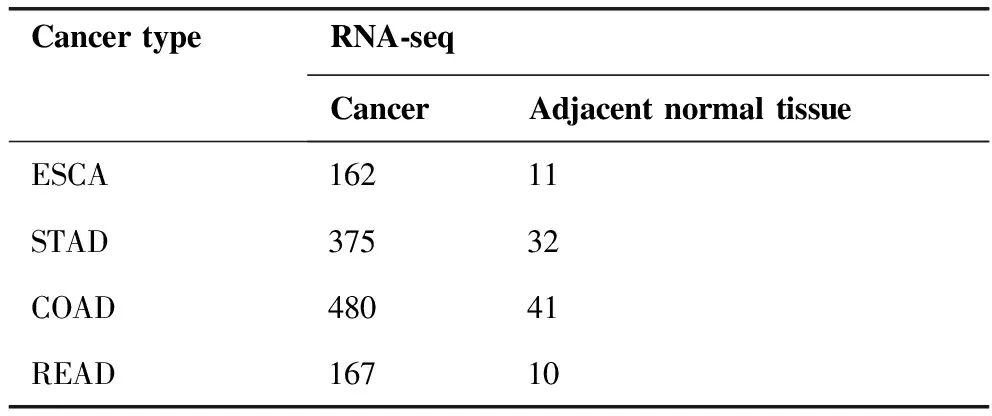
表1 消化道癌症RNAseq数据基本信息
1.2 差异基因的筛选
首先,本研究分别构建每个消化道癌症RNAseq表达谱,利用R软件包里的DESeq2软件包(1.34.0版)[10]在R软件上进行差异分析。其次,以|log2FC|>1.5和P<0.05作为筛选差异基因的阈值选取差异基因,分别得到每种消化道癌症的差异基因。最后,通过对这三类癌症的差异结果取交集,获得共享差异基因,并区分共同上调和共同下调的基因。火山图及韦恩图分别用于展示差异结果及共享的差异基因。
1.3 功能富集及通路富集分析
利用R软件包里的clusterProfiler软件包(4.2.0版)分别对差异基因、共同上调基因及共同下调基因进行GO功能富集及通路富集分析[11]。GO富集分析基于超几何分布,应用超几何检验进行富集分析,利用Benjamini-Hochberg方法对富集结果进行P值校正,以校正后的P<0.05作为标准选择差异显著的富集结果。
1.4 蛋白互作网络分析
STRING[12]数据库(八版)包含人类蛋白之间的互作信息,本研究通过STRING数据库获取差异基因蛋白互作信息,并由Cytoscape软件3.8.0版进行蛋白互作网络的可视化,并借助软件Cytoscape中的cytoNCA插件(2.1.6版)进行网络拓扑属性分析,设置条件为:Betweenness和degree项均选without weight。以此为基础筛选蛋白互作网络中具有高连通度的节点。
利用Cytoscape软件里的mCODE插件(1.6.1版)对蛋白互作网络进行分解,设置条件为:node score cutoff项选0.2;K-Core项选0.2;Degree Cutoff项选2;max.Depth项选100。得到的模块将被用于进一步的分析,分析这些子模块里面的节点及其邻居节点,分析密集且重于的互作的模块的拓扑属性并对核心节点进行拓扑学分析。
1.5 转录因子及靶基因的识别
TRRUST数据库2.0版是一个包含转录因子及其调控的靶基因信息的在线数据库,通过在TRRUST在线数据库查询并识别消化道癌症共享的差异基因中的转录因子及转录因子调控的差异基因。
1.6 子模块的分析
为了更深入的了解这些转录因子在转录前水平基因转录调控的作用及其靶基因到下游的基因表达水平的影响,结合TF-mRNA调控信息及蛋白互作通过绘图分析TF及TF所调控的基因对下游表达水平的影响。
2 结 果
2.1 差异基因筛选
通过差异分析之后进行取交集本研究识别到食管癌、胃癌,直肠癌及结肠癌之间共享的差异基因共有741个,其中共同上调的基因有333个,共同下调的基因有337个,如图1所示,四种癌症之间的差异结果用火山图进行展示如图1,其中四个癌症之间的共享基因则用韦恩图将来进行展示,如图2。
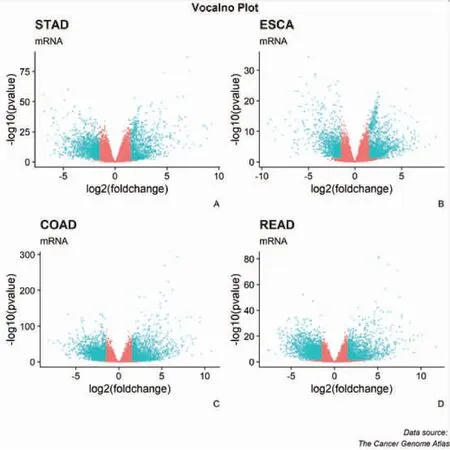
图1 四种消化道癌症差异基因火山图
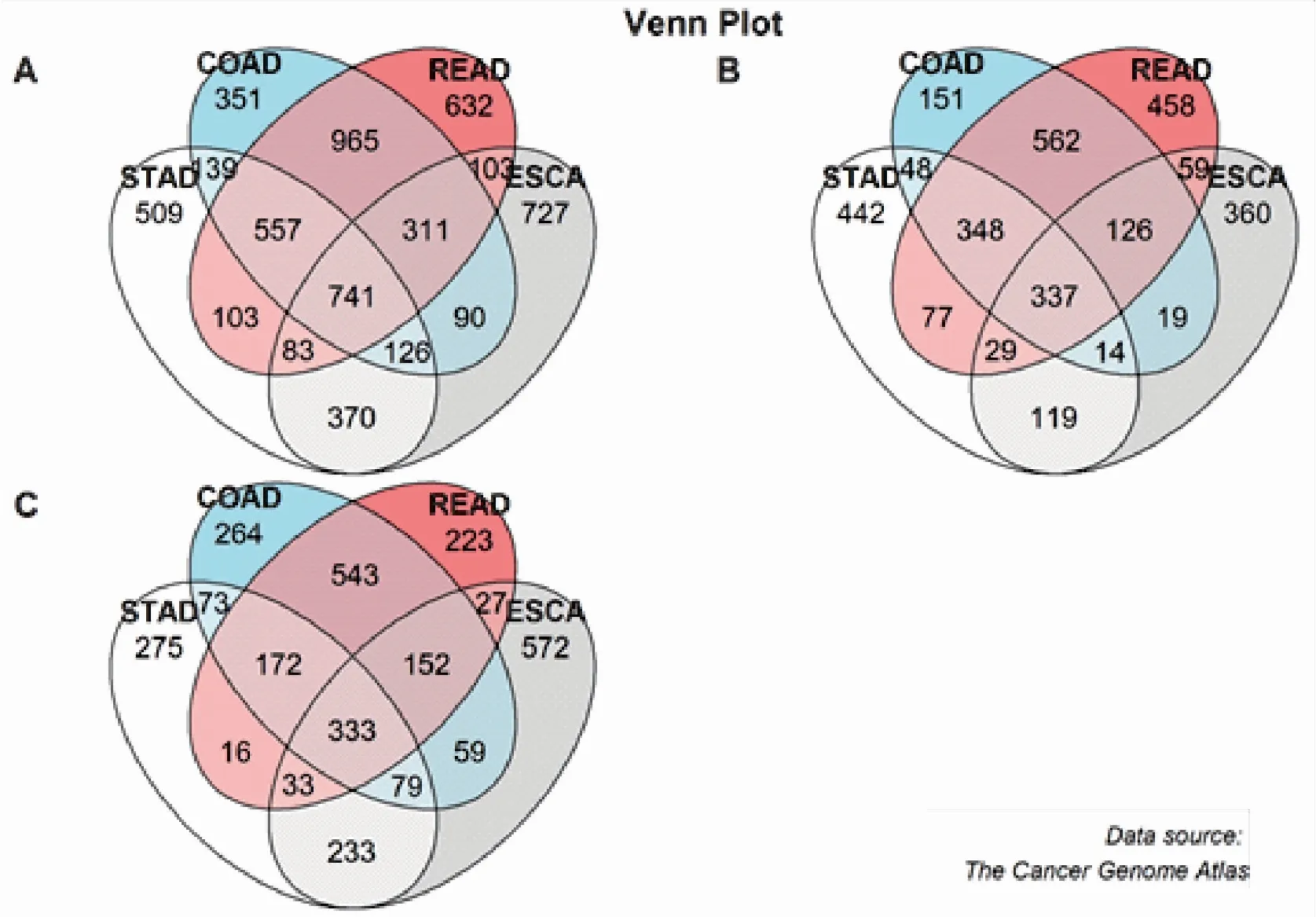
图2 四种癌症的共享基因韦恩图
2.2 功能富集分析以及通路富集分析
如图3所示,差异基因基因功能主要富集在肌肉收缩(muscle contraction)、肌肉系统过程(muscle system process)、胞外基质形成(extracellular matrix organization)、胞外结构形成(extracellular structure organization)等功能。而共同上调基因主要富集的功能有胞外基质的形成(extracellular matrix organization)、胞外结构形成(extracellular structure organization)等功能,下调基因主要富集在肌肉收缩(muscle contraction)、肌肉系统过程(muscle system process)及膜电位的调节(regulation of membrane potential)。
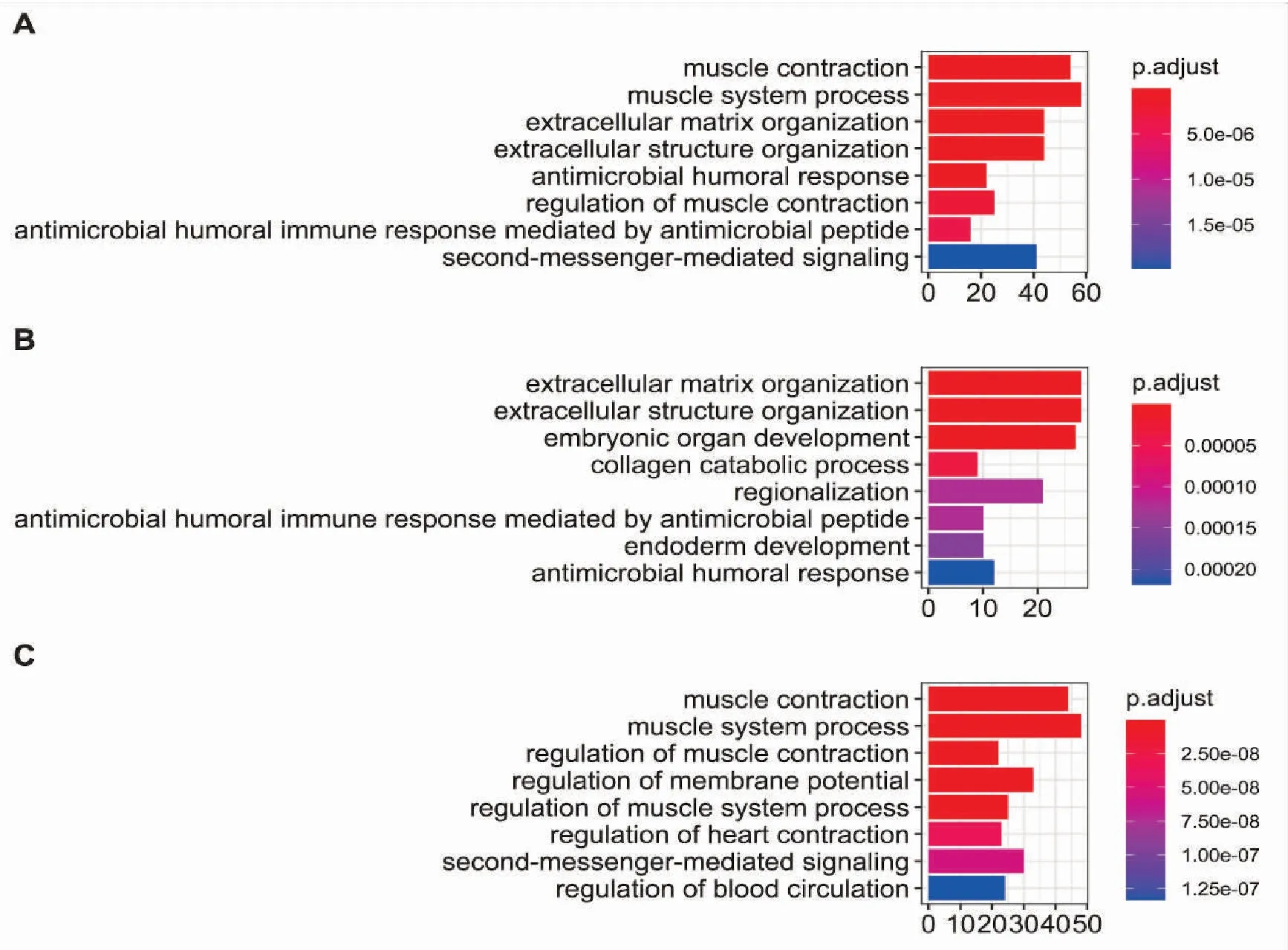
图3 功能富集分析图
如图4所示,通路富集分析显示差异基因主要参与的通路有GPCR配体结合(GPCR ligand binding)、类A/1(视红紫质样受体)(Class A/1(Rhodopsin-like receptor))等通路。共同上调基因主要参与了肽配体结合受体(Peptide ligand-binding receptors)、胶原降解(collagen degradation)及由胰岛样生长因子结合蛋白介导的胰岛样生长因子的转运及吸收(regulation of insulin-like growth factor(IGF)transport and uptake by insulin-like growth factor binding proteins(IGFBPs))、转录前蛋白磷酸化(post-translational protein phosphorylation)等通路,共同下调基因参与的通路有神经系统(neuronal system)、神经递质受体和突触后信号传输(Neurotransmitter receptors and postsynaptic signal transmission)、平滑肌收缩(smooth muscle contraction)、离子稳态(ion homeostasis)。
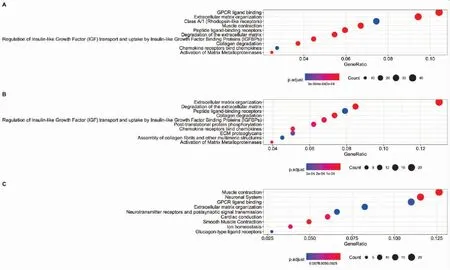
图4 通路富集分析图
2.3 蛋白互作网络分析
借助STRING蛋白互作数据库,获取741个差异基因的蛋白互作信息,并用Cytoscape进行网络绘图,发现蛋白互作网络共有731个节点,3 038条边。
利用CytoNCA插件针对连通度(Degree),介数(Betweenness),紧密度(Closeness)对网络中的核心节点进行分析,部分网络节点属性如表2。以Degree>25为标准筛选核心节点,共别到52个基因如CXCL8,SPP1,CXCL12等。
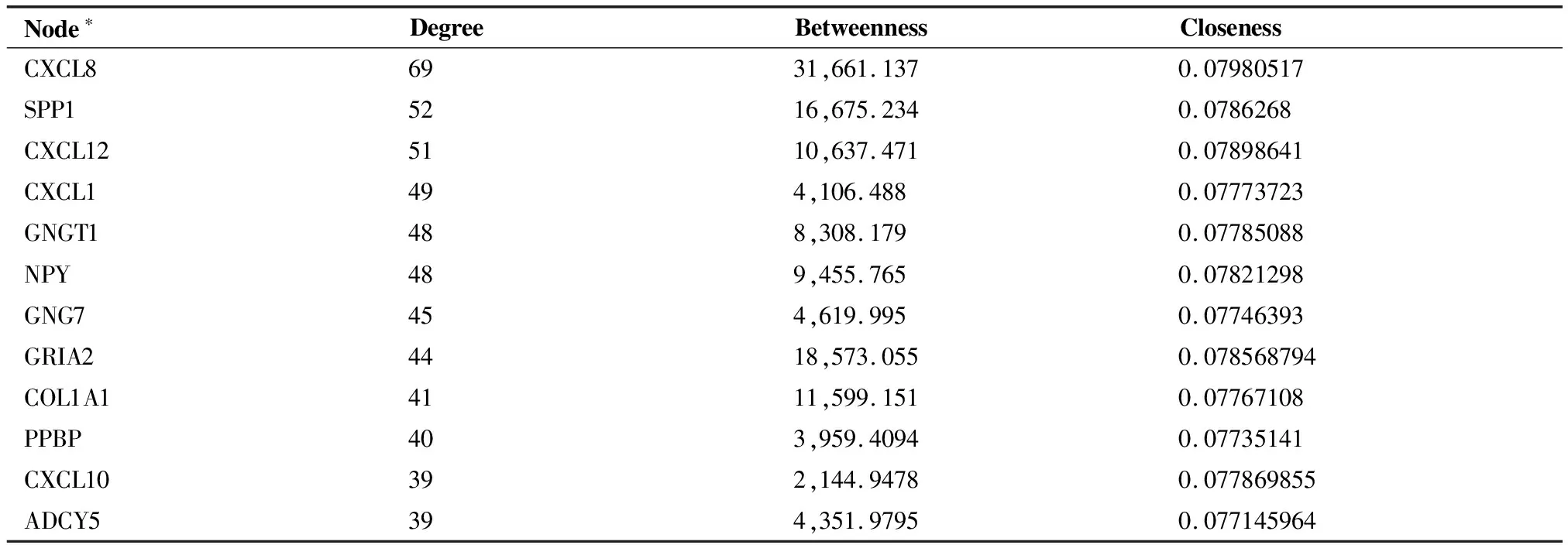
表2 蛋白互作网络节点的属性
利用软件Cytoscape里的MCODE插件,对蛋白互作网络进行分解网络分解,获得29个模块,部分模块分析结果如表3。对结果中前四个Module用R软件包里的igraph软件包1.2.11版进行绘图,如图5所示。
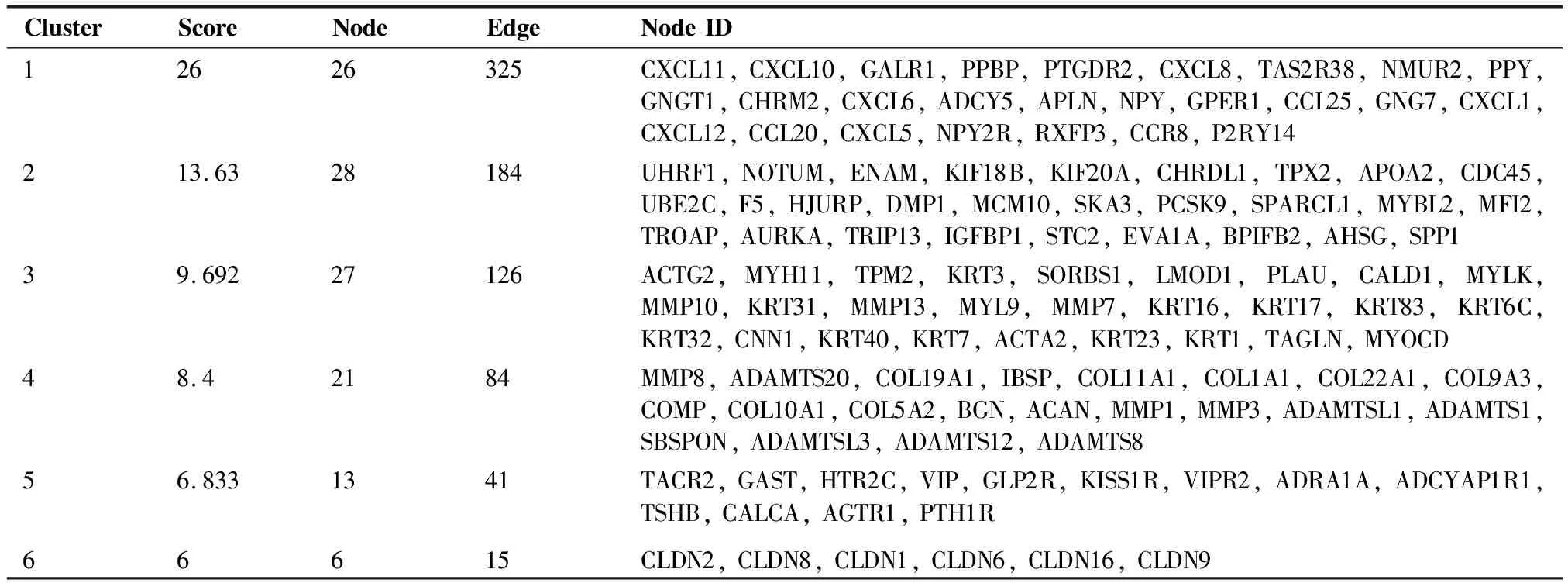
表3 网络模块分析结果
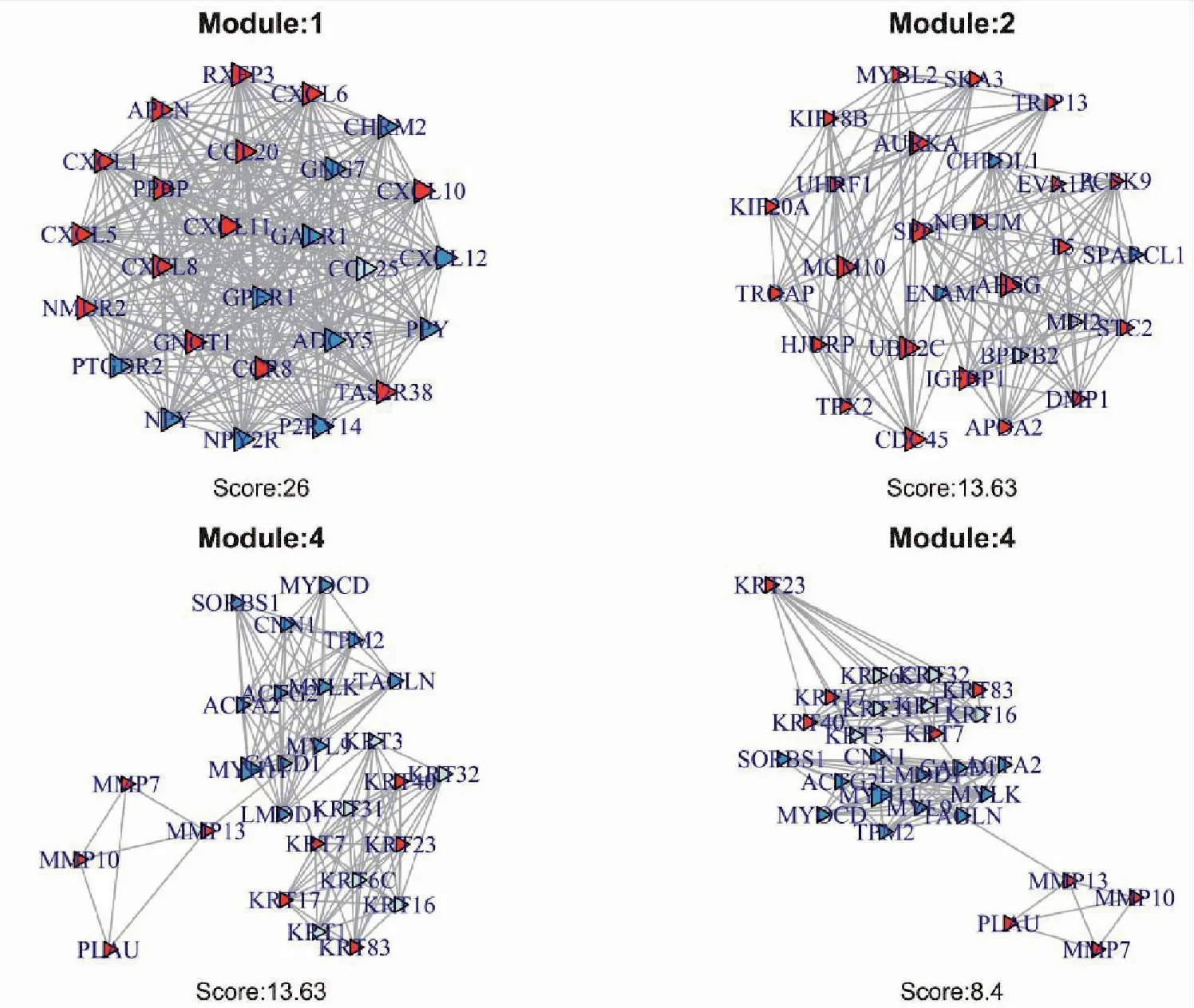
图5 网络模块
2.4 转录因子及其靶基因的识别
本研究借助TRRUST在线数据库,对差异基因中的转录因子进行预测。识别在差异基因中起到转录因子作用的基因共有13个,最终识别到28对TF-mRNA调控关系及部分调控方式诸如激活和抑制,调控关系预测结果如表4所示。

表4 TF-mRNA 调控关系
2.5 子模块分析
结合TF-mRNA调控信息及蛋白互作网络模块结果,分析TF所调节的基因所参与的功能。如图6 Module 2所示,在所有的上调基因中有TF基因OTX2调控AURKA,AR调控CDC6及MYBL2调控MYBL2。特别指出,AR除了作为转录因子调控CDC6之外,也作为基因与CDC6存在蛋白互作关系。结合表4中TF-mRNA调控信息及基因本身表达水平,分析TF及TF所调控的基因的在模块中起的作用。
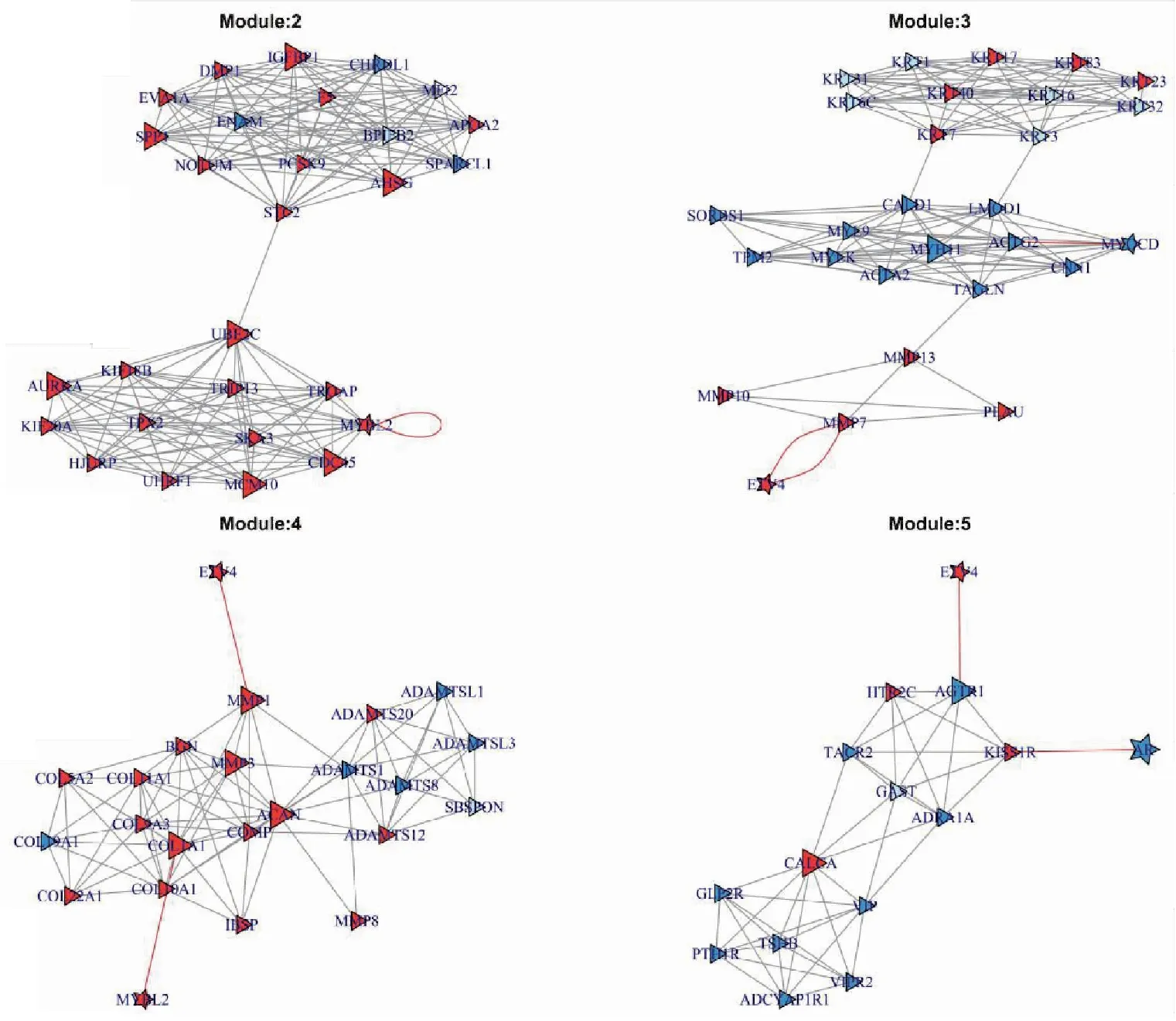
图6 子模块分析
3 讨 论
消化道癌症包括食管癌、胃癌、结肠癌及直肠癌,这四种癌症都对人群健康造成了巨大的威胁。从组织胚胎发育起源的角度分析,消化道癌症之间有共同的内胚起源,且都暴露于相似的导致癌症形成的管道环境致病因素[13]。消化道在消化系统中起着营养消化吸收等重要功能,消化道组织学上的癌变情况影响到原先正常的消化道的组织结构乃至原先的正常功能。癌变的过程是一个复杂的过程,癌症肿瘤结构的改变到在病理上的改变往往是从功能学上的直至出现器质性改变。出于这两个立足点出发,分析消化道癌症之间的共享的分子特点,本文系统分析四种消化癌症间共享功能,其中结果提示主要富集在肌肉收缩、肌肉系统过程、胞外基质形成、胞外结构形成等功能,这些功能差异最为显著的是肌肉收缩、平滑肌收缩,其中,肌肉收缩的功能在消化道癌症中的起着极其重要的功能,本文功能富集分析结果发现消化道癌症中下调基因显著地富集于肌肉收缩等功能,表明癌变的消化道肌肉收缩功能的显著下降,而在上调基因富集结果表明胞外基质形成、胞外结构形成及胶原代谢等功能,已有Wu等[14]研究指出I型胶原在结肠癌中通过整联蛋白α2β1介导PI3K/AKT信号通路增强结肠癌干细胞的特性和强烈的侵袭性。
通过分析蛋白互作网络的拓扑属性如连通度等属性,本研究筛选出52个具有高连通度的基因如CXCL8,SPP1,GNGT1,GRIA2等。基于表达水平,发现CXCL8,CXCL1,CXCL10,CXCL5,PPBP,WT1,PAX2等31个在三个癌症中表达均上调,而在共同下调的基因中如SPP1,ADCY5,CXCL12,PTGDR2则呈下调。其中发现在上调基因中存在CXC家族的基因有CXCL1,CXCL5,CXCL6,CXCL8,CXCL10,CXCL11,如CXCL8及CXCL12,均被相关研究指出CXCL基因主要参与趋化因子受体结合趋化因子等功能且都具有可作为胃癌预后的生物标志物[15-17]。结合功能富集分析结果显示,CXC家族基因主要参与涉及细胞因子信号传导等功能。除CXC家族基因外,其他上调基因基因如MMP1通过P13K/AKT通路促进食管鳞状细胞癌的发生[18]。这些基因在功能上形成一些功能结合体如功能模块行使特定的功能,这些模块里面的基因在功能层面上紧密连接,对功能的维持极其重要。
本研究共识别到13个转录因子如AR,CBX7,ETV4,HOXC6及HOXC8等,其中存在转录调节关系的共有28对,如AR调节KISS1R、PEMPA1、UTG2B15及USP26。对蛋白互作网络的结构在连通度大于25且受到转录因子调控的基因有COL1A1,MMP1等。其中在本研究预测结果中的转录因子中,其中PAX2和WT1不仅在蛋白互作网路中作为高连通度的节点,且作为转录因子在转录前对靶基因进行表达调节。其中转录因子如ETV4已被发现在消化道基底癌中通过经典Wnt/β-catenin通路影响细胞周期[19]。其他转录因子如HOXC6亦被报道通过活化剂protein-1通路在消化道癌症影响肿瘤的生长[20]。其中MYBL2调控COL1A1,其中COL1A1被发现与肿瘤转移性质有关,所以MYBL2作为重要转录因子在结肠癌中起重要作用[21],但目前并没有相关实验证据证明其他消化道癌症如胃癌被报道与MYBL2的作用与效应,MYBL2在胃癌及食管癌中的表达作用及参与的重要通路仍需进一步深入分析。
结合模块分析结果TF-mRNA转录调控关系,分析转录因子及其所调控的基因起的调节作用,模块2中,MYBL2作为转录因子调节MYBL2,MYBL2作为模块2中的分子又与其他基因存在蛋白互作关系。模块3中,ETV4对MMP4存在两种调节关系,因为模块本身是由于基因之间紧密的作用形成的一种集合,任何对其中模块中的节点的影响都间接地影响到模块中的其他基因。最终识别重要的转录因子AR,CBX7,ETV4,HOXC6及重要的基因CDC6,NCAM1,AGTR1,MMP1,COL1A1。为深入研究消化道癌症间共享的机制及转录前TF调节机制重要分子提供深入的分子层面的并为消化道癌症之间的治疗提供启发。
4 结 论
本研究通过构建TF-mRNA调节网络,发现消化道癌症之间重要的共享功能有肌肉收缩、肌肉系统过程、细胞外基质的组织、构建细胞外结构,并识别到重要的转录因子AR,CBX7,ETV4,WT1, PAX2及基因CDC6,NCAM1,AGTR1,MMP1,COL1A1。为消化道癌症间识别到重要的共享转录因子及靶基因,为开发更安全更有效的药物提供更好提供理论支持。
作者声明:本文全部作者对于研究和撰写的论文出现的不端行为承担相应责任;并承诺论文中涉及的原始图片、数据资料等已按照有关规定保存,可接受核查。
学术不端:本文在初审、返修及出版前均通过中国知网(CNKI)科技期刊学术不端文献检测系统的学术不端检测。
同行评议:经同行专家双盲外审,达到刊发要求。
利益冲突:所有作者均声明不存在利益冲突。
文章版权:本文出版前已与全体作者签署了论文授权书等协议。
猜你喜欢
理财周刊(2022年4期)2022-04-30
中国饲料(2022年5期)2022-04-26
科学导报·学术(2020年47期)2020-11-17
学苑创造·A版(2020年12期)2020-01-07
中国保健营养(2019年9期)2019-10-20
学习与科普(2019年4期)2019-09-10
商情(2012年48期)2017-11-08
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年15期)2017-08-30
江苏农业科学(2017年5期)2017-04-15
