
萃取过程中微观到宏观的多尺度超分子组装
——离子液体的特异性功能
2022-03-14 06:49沈兴海
核化学与放射化学 2022年1期
史 策,沈兴海
北京分子科学国家研究中心,放射化学与辐射化学重点学科实验室,应用物理与技术研究中心,北京大学,化学与分子工程学院,北京 100871
液-液萃取作为一种最为传统和成熟的分离方法,在金属离子的分离与富集中有着广泛的应用[1-8]。传统的液-液萃取化学通常假定萃取剂和萃合物在萃取有机相中以单分子的状态存在,探究金属离子配位作用和水合作用等对萃取机理和效率的影响,较少考虑两相中萃取剂和萃合物分子的聚集状态[1]。但实际上萃取剂和萃合物可能具有较强表面活性,萃取体系中可进一步形成多种超分子聚集体,如胶束、囊泡等[2-6]。本课题组综述了反相胶束与微乳液萃取分离金属离子的最新进展[2]。这些超分子组装过程通常均局限于微观或介观尺度。最近,本课题组发现在离子液体萃取体系界面形成宏观超分子组装(macroscopic supramolecular assembly, MSA)新现象[9-10],表明萃取体系中的超分子组装可以进一步发展至宏观尺度。这为研究溶剂萃取机理,进而开发新的分离方法和流程提供了新的思路。
液-液萃取是一个界面相互作用的过程。由于萃取体系的界面行为复杂,缺少有效的研究手段,所以萃取体系的界面性质和界面行为还有待进一步研究。目前,已经有工作发现,在金属离子的萃取过程中,金属离子与萃取剂分子之间的相互作用发生在液-液界面处[7-8]。显然,深入分析萃取体系界面性质和萃取过程中金属离子、萃取剂分子的界面行为是一种十分直接而本质的研究策略。随着表征手段和计算方法的不断发展,从界面角度研究液-液萃取过程逐渐成为新热点[11-13]。
离子液体(IL),是一种由阴阳离子构成、在室温或接近室温(<373 K)下呈现液态的有机盐类化合物,又称室温离子液体[14]。离子液体本身具有许多非常突出的优点:(1) 化学稳定性和热稳定性高;(2) 蒸气压低;(3) 电导率高,电化学窗口宽;(4) 溶解能力强;(5) 可通过设计不同阴阳离子,合成不同物理化学性质的离子液体。离子液体代替普通有机溶剂时,萃取体系充分体现了离子液体的优点,萃取流程绿色环保,且具有高萃取效率、高选择性和高辐照稳定性等优点[15]。此外,离子液体的独特结构能够使萃取分离过程发生复杂的超分子组装行为,并可在界面处实现MSA[9,16]。目前,已有综述分析了离子液体体系的萃取行为及其在乏燃料后处理中的应用前景[15,17]。然而离子液体本身黏度大,且萃取过程中易流失,所以还需考虑提高传质速率和离子液体的损失与复用等问题[2]。
本文主要围绕萃取过程中微观到宏观的不同尺度超分子组装行为进行评述,对界面性质、离子交换行为以及介观尺度超分子组装体的结构进行分析总结,重点介绍实现萃取体系MSA的方法及其分离性能。MSA过程中界面上呈现出持续的Marangoni效应,推动了MSA的进行。本文着重介绍离子液体萃取体系,离子液体可与介质发生多种非共价相互作用,从而产生丰富的超分子组装行为,MSA即是在离子液体萃取体系中实现的。
1 萃取过程微观界面性质和离子转移
Choppin等[18]总结了金属离子配位和水合作用对溶剂萃取分配比的影响和萃取剂的结构对萃取热力学、动力学平衡的影响。萃取过程中,水相中的溶质通过界面区域转移到另一相,液-液界面的能量势垒会对其造成显著影响[19-20]。因此,研究萃取体系的界面性质与离子转移过程具有重要意义。本部分主要介绍萃取体系的微观界面性质和跨越界面的离子交换行为。
1.1 萃取体系微观界面性质
液-液萃取是一个界面相互作用的过程。由于萃取体系的界面行为复杂,缺少有效的研究手段,所以对萃取体系的界面性质和萃取过程的界面行为还有待进一步研究。近年来,随着界面的实验表征手段和理论计算方法的不断发展,从界面角度研究萃取过程逐渐成为了液-液萃取研究的新方向。
为了研究萃取体系的界面行为,采用了大量的界面表征手段。如全内反射荧光光谱(TIRF)[21]、振动和频光谱(SFG)[22-24]、二次谐波方法(SHG)[11-13]、中子反射谱[19-20]、全内反射紫外吸收光谱(TIR-UV)[25-26]、衰变全反射光谱[27]、扫描离子电导显微镜[28]等。Richmond课题组[22]通过SFG技术分析了多种有机溶剂和水的界面,通过对比水分子在不同界面环境中的SFG信息,解析了水分子在液-液界面上的排布规律。Ishizaka等[21]通过TIRF分析了分子在液-液界面上的各向异性。有机溶剂改变,液-液界面平整度改变,导致分子各向异性也有所不同,由此得到了界面厚度等相关信息。Martin-Gassin等[11]用SHG研究了酰胺类萃取剂N,N-二(2-乙基己基)异丁酰胺(DEHiBA)的萃取过程。DEHiBA具有表面活性,可吸附在水/油界面。SHG信号可原位检测界面处萃取剂与离子、分子的相互作用,从而定性分析其界面分布行为。Scoppola等[19-20]将X射线反射谱和中子反射谱结合起来,研究了二酰胺类萃取剂N,N-二甲基-N,N′-二辛基己基乙氧基丙二酰胺(DMDOHEMA)萃取Nd3+体系中水/油界面结构。X射线反射谱可分析水相中的Nd3+,中子反射谱可检测有机相中的DMDOHEMA。由此实现了该萃取体系各物种在界面上分布行为的定量分析。Sun等[25-26]通过TIR-UV监测了Cr3+在水/油界面的吸附行为,并通过在界面处的吸光度变化分析了Cr3+与其他离子的竞争吸附行为。该测试是在其自制的石英反射槽中进行,如图1所示,紫外信号光通过棱镜折射,在液-液界面处发生反射,最后再经过棱镜折射进入信号收集系统,从而得到TIR-UV结果。通过TIR-UV特征峰的强度来表征Cr3+在界面处的浓度。
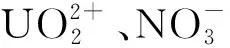

(a):1代表石英棱镜,2代表石英槽图1 用于TIR-UV测量的样品槽的结构示意图(a)和不同浓度铬酸钠溶液/CH2Cl2溶液界面处的TIR-UV(b) [25]Fig.1 Schematic of the experimental set-up of the reflection quartz cell used in the TIR-UV measurements(a), and TIR-UV spectra of Na2CrO4 solutions at different concentrations in the CH2Cl2/H2O interface(b)[25]

(a)——二价离子与两个DEHP分子作用,(b)——三价离子与三个DEHP分子作用黑色实线表示液-液界面;该图解释了配位作用造成的液-液界面曲率图2 二价(a)与三价(b)金属离子分别与DEHP配体在液-液界面发生配位作用的示意图[8]Fig.2 Schematic diagrams of the interaction of the extractant DEHP with a divalent ion(a) and a trivalent ion(b) at the liquid-liquid interface[8]
综上可以看出,通过研究萃取体系的界面性质和萃取剂、金属离子的界面行为,有助于理解金属离子跨越界面的微观机理,进而设计并开发新的策略来提升萃取的分离效率和选择性。
1.2 萃取过程的离子转移行为
在萃取过程中,离子在液-液界面上的转移反应是其基本的物理化学过程之一,可以通过电化学的方法研究电荷转移现象,实现离子转移动力学过程的实时监测。另外,采用电化学方法可以将界面减小至纳米尺度,排除其他外在因素对离子转移过程的干扰[32]。
微/纳米玻璃管支撑的液-液界面研究可揭示其快速电荷转移反应的过程。Li等[33]研究了四乙基铵离子(TEA+)在水/1,2-二氯乙烷(DCE)界面的简单离子转移反应,其反应速率常数会受到玻璃管径影响,当其半径小于5 nm时,反应速率常数最高可达(110±23) cm/s。多离子物种在两种不混溶电解质溶液界面上的离子转移行为也可通过电化学手段进行研究。Li等[34]研究了第一代聚酰胺亚胺树枝状高分子G1的离子转移过程。通过循环伏安法观察到树枝状分子与萃取剂二苯并-18-冠-6(DB18C6)之间存在主客体相互作用,转移电位发生了负位移。从树枝状分子G1的离子转移行为,发现每个DB18C6分子可以选择性地与一个氨基配位。并利用电化学质谱联用技术准确评估了G1在界面上转移时存在[DB18C6-G1-DB18C6]2+这种转移中间体,为中性萃取剂加速生物大分子的输运提供了借鉴意义。
电化学手段也可研究萃取过程中金属离子与萃取剂形成的配离子结构。Stockmann等[35]分别研究了Sr2+与正辛基苯基-N,N′-二异丁基胺甲酰甲基氧化膦(CMPO)在水/DCE和水/离子液体微界面上的配位作用,发现在水/DCE微界面主要存在 [Sr(CMPO)2]2+和[Sr(CMPO)3]2+两种配离子,而在水/离子液体微界面形成的配合物只有[Sr(CMPO)3]2+一种。这也解释了相比于烷烃,离子液体体系萃取效率更高的原因。其他金属离子的界面转移行为也可通过类似方法研究。最近,Jiang等[36]利用脉冲伏安法分析了Fe3+与四种不同配体在水/DCE界面的结合常数,并计算了其结合的化学计量数之比,由此筛选出萃取效果最优的配体。
1.3 新型离子液体萃取体系的微观界面性质与离子转移
如引言部分所述,离子液体萃取体系因其高萃取效率、高选择性和良好的辐照稳定性而受到广泛关注。离子液体的界面性质与结构可显著影响多相体系反应和分离过程的效率,成为了重要的科学问题[37-40]。另外,离子液体萃取体系复杂的界面结构也给研究萃取过程中的离子转移机理带来了新的挑战[15]。

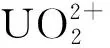

图3 钍离子的萃取-反萃过程示意图(a)和超临界CO2从离子液体相反萃Th(Ⅳ)装置图(b)[51]Fig.3 A diagram showing the extraction-stripping procedure for thorium(a) and apparatus for the stripping of Th(Ⅳ) from the IL phase to SC-CO2(b)[51]
2 萃取体系介观尺度的超分子组装
萃取过程中,一些萃取剂与金属离子结合生成配合物。在此基础上萃取体系可以进行进一步的超分子组装,进而聚集形成反相胶束。随着金属离子浓度的提高,反相胶束的结构可能改变甚至发生二次组装。随着研究的推进,人们对萃取体系中形成的囊泡、胶束等介观超分子聚集体有了更深的认识。在本文中,介观尺度一般界定为100 nm到1 000 nm之间,100 nm以内的为微观尺度,微米及微米以上为宏观尺度[59-60]。本部分首先介绍萃取过程中介观尺度的超分子组装体状态,之后介绍离子液体体系中的超分子聚集体。
2.1 有机溶剂萃取体系介观超分子组装


图4 POP5A在C8mimNTf2体系中对选择性萃取的示意图(插图:POP5A结构)[58]Fig.4 Schematic of selective extraction on in POP5A/C8mimNTf2 system(The structure of POP5A is shown in the inset)[58]


图5 HDEHP萃取Th4+后形成棒状胶束和蠕虫状胶束示意图[65]Fig.5 Schematic diagrams of rod-like micelles and worm-like micelles formed after the extraction of Th4+ by HDEHP[[65]
2.2 离子液体萃取体系中的超分子聚集体
离子液体参与形成的超分子体系也成为近年来研究的热点。离子液体可以构筑微乳液体系,并用于萃取[2,69],而在混合体系中被广泛研究的离子液体一般具有亲水的极性头基和疏水的烷基侧链,具有表面活性,可在多种溶剂体系中聚集形成不同的超分子组装体,如胶束、囊泡、溶致液晶等[16]。
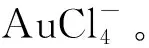

图6 自组装囊泡形成及囊泡提取Au的工艺示意图[73]Fig.6 Schematic illustration of the formation of self-assembled vesicles and the process of gold extraction by the vesicles[73]

图7 由DGA2-及形成的囊泡(a)和连接囊泡(b)示意图[74]Fig.7 Schematic diagram of the vesicles formed by DGA2-and induced link of the vesicles(b)[74]
在离子液体萃取体系中构筑反相胶束或囊泡体系以进行分离,可以避免使用传统有机溶剂,充分体现了离子液体的优势。
3 萃取过程中宏观尺度的超分子组装行为
通常,萃取体系的组装最终都停留在了介观尺度。最近,本课题组和中国工程物理研究院[9]共同报道了离子液体萃取体系界面上的MSA行为,并实现了方便快捷的分离。本部分首先简介MSA的研究概况,之后着重介绍离子液体萃取体系界面上的MSA。体系界面呈现出持续的Marangoni效应(Marangoni效应是指体系在外界扰动下产生了局部界面张力梯度后,分子自发向高表面张力的方向移动的过程),在MSA过程中发挥了重要作用。
3.1 宏观超分子组装简介
一般地,将构筑基元尺寸在微米及以上尺度的自组装统称为宏观组装(macroscopic assembly, MA)[59,75-76],当构筑基元进入微米乃至毫米、厘米尺度时,布朗运动已经难以驱动宏观构筑基元发生有效碰撞。所以要实现宏观组装,则需要引入新的驱动力。同时,考虑到构筑基元尺寸较大,远大于分子间作用力的作用距离。早期的研究主要采用引入的作用力程与构筑基元尺寸匹配的方案,即利用毛细作用力[77-79]、电场力[80-82]、磁场力[83-85]等作用力程可达厘米范围的长程作用力来实现宏观组装。但其缺陷在于外界环境如界面、电场、磁场等有着较强的依赖性,离开这种特定的外界环境后,组装体会发生解体,再次回到构筑基元的状态。针对这个问题,人们选择引入超分子作用来形成稳定的组装体,即MSA[75]。
宏观尺度构筑基元的表面通常并非完美表面,一般会比较粗糙,其粗糙程度甚至远远大于超分子作用的力程范围。针对宏观构筑基元表面粗糙的问题,Harada等[86]通过在水凝胶体系中引入具有分子识别的主客体官能团,首次实现了MSA。他们通过机械振荡使水中修饰了β-环糊精的主体水凝胶块和修饰了金刚烷的客体水凝胶块发生碰撞,两种水凝胶块一经接触即可形成一体,从而实现水凝胶的MSA。除了主客体识别作用外,该课题组还进一步引入金属配位作用[87]、氧化还原作用[88]、碱基配对[89]等,分别实现了宏观凝胶间的超分子组装与解组装。
这一过程中构筑基元之间仍依赖振荡或旋转等方式迫使其相互碰撞从而发生组装,但这种无规地、随机地碰撞就会使得组装体有序程度较低[90]。Xiao等[91]设计的宏观构筑基元内部预留空腔用来装载低表面能的乙醇。如图8所示,当把该构筑基元置于空气/水界面上时,乙醇被释放并接触水,这会在表面局部形成表面张力梯度,从而在Marangoni效应驱使下发生构筑基元的自发移动并预先调节构筑基元匹配程度。另外,构筑基元表面修饰有超分子主客体识别层,使得构筑基元在发生长程精准匹配之后,通过主客体作用形成稳定的组装体。Cheng等[92]进一步在水相引入超分子识别体系。构筑基元释放的表面活性剂分子可以与溶液中的β-环糊精发生主客体作用,使得表面活性剂在界面上达到饱和吸附的时间增长,从而将Marangoni效应的持续时间从120 s延长至了2 200 s。虽然有所改善,但Marangoni效应持续时间依然有限,导致MSA过程不能持续进行。
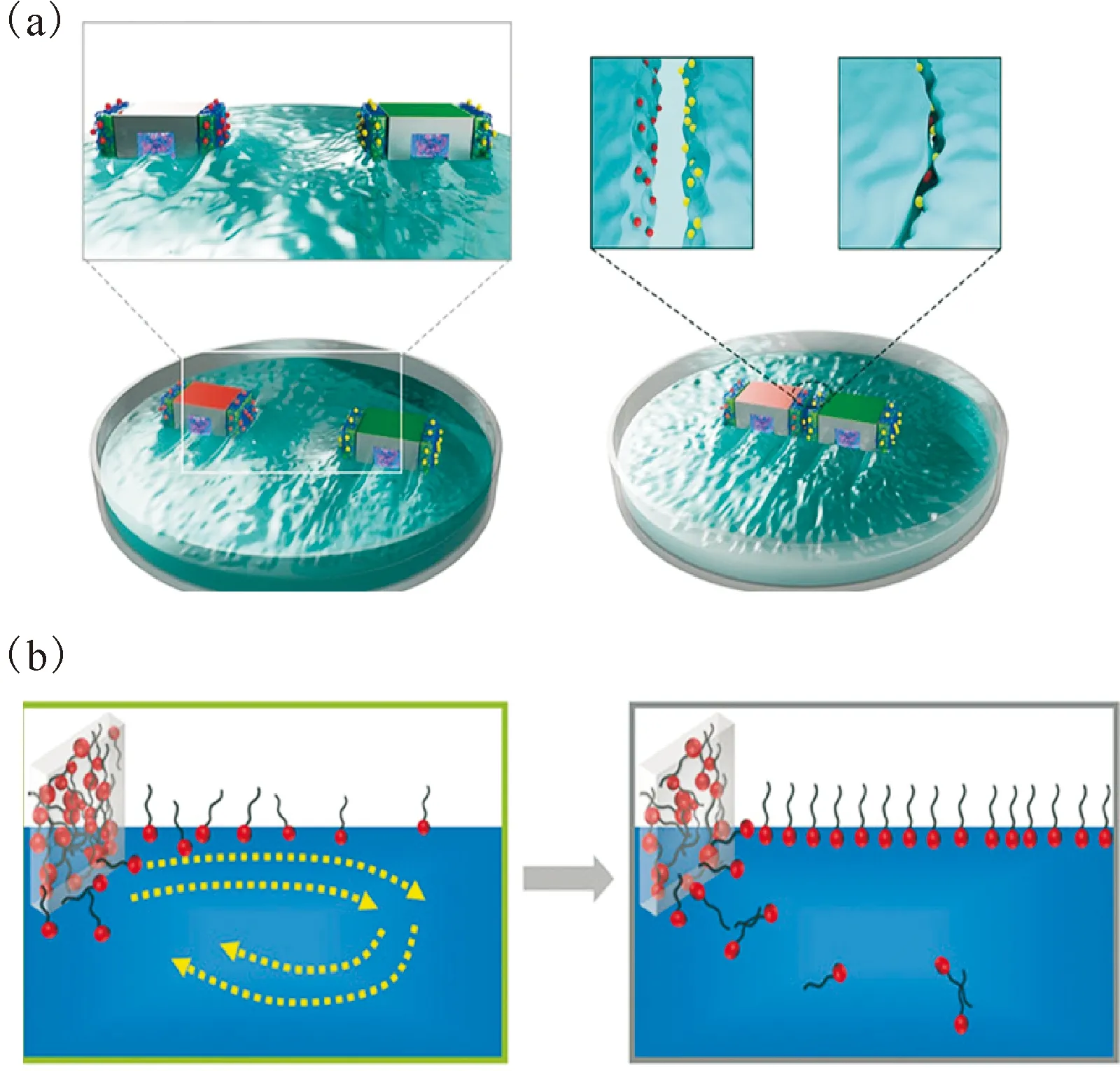
图8 自发的MSA过程示意图(a)和Marangoni效应的作用机制示意图(b) [92]Fig.8 Schematic of the process of spontaneous MSA(a) and the mechanism of Marangoni effect(b)[92]
3.2 离子液体萃取体系中的宏观超分子组装
受到上述工作[91-93]的启发,考虑到离子液体本身具有较强的表面活性[40],如果能够在离子液体萃取体系中,使得在界面上因界面张力梯度而呈现出持续的Marangoni效应[9-10],则有望实现萃取体系中的MSA。

图9 基于宏观超分子组装的铀的萃取方法(a)和宏观超分子自组装机理示意图(b)[9]Fig.9 Extraction based on MSA of the and the potential formation mechanism of uranium-rich MSA sample(b) [9]
进一步研究了MSA的结构和形成机理。萃取过程的化学反应平衡方程式[94]为:
(1)

(2)
该工作不仅丰富了离子液体体系中的萃取方法,而且为MSA领域的一个重要科学问题即持续推动力问题的解决,提供了新的途径。基于羟基离子液体特殊的表面性质及其参与阳离子交换并与萃取剂和铀酰离子配位造成局部浓度变化这一特点,在界面创造了连续的Marangoni效应,解决了其持续时间较短的难题,进一步实现了铀与配体螯合物从分子尺度到介观尺度再到宏观尺度的自组装,整个过程没有引入任何外力,是自发的热力学过程。
在上述工作基础上,本期离子液体专栏中还报道了基于离子液体萃取体系MSA的锶分离研究[10]。结果表明,可通过离子液体萃取体系界面的MSA实现Sr2+的选择性分离和提取。可实现MSA的离子液体为CnOHmimNTf2(n=2、3),萃取剂为CMPO和N,N,N′,N′-四辛基-3-氧戊二酰胺(TODGA)。该MSA过程机理为与U-MSA类似的多级超分子组装机理。该分离系统能够成功实现Sr2+、Cs+混合水溶液中Sr2+的一步法提取和固化,且同样不需要反萃,形成的Sr-MSA样品可方便快捷地取出。该方法在高酸度场景如乏燃料后处理中具有潜在的应用价值。
4 总结与展望
本文对萃取体系中的超分子组装行为进行了较系统的总结。文献中已经报道了很多萃取体系中的超分子组装行为,但这些研究大都集中在了微观和介观尺度。目前萃取体系中的MSA已经得到报道,并由此实现了方便快捷地分离。该工作有待进一步研究。一方面,可以继续扩展针对萃取体系MSA过程的分离应用。该方法的选择性、放射稳定性等问题有待进一步研究,并可针对具体场景如乏燃料后处理提出基于MSA的概念性分离流程。乏燃料后处理或其他放射性的应用场景中,传统的萃取往往伴随着大量放射性有机废液需要处置。基于MSA的分离方法可直接实现金属离子的固化,无需反萃,这将对分离科学的发展具有重要意义。另一方面,深入研究MSA体系界面上持续的Marangoni效应,可以为MSA领域的一个重要科学问题即持续推动力问题,提供新的解决途径。
萃取体系的界面十分复杂。随着界面表征手段和计算方法的发展,目前已经有文献报道了金属离子与萃取剂在界面处的作用机制。金属离子和萃取剂被吸附在水/油界面上,并在界面处形成萃合物,进一步地萃合物会脱离界面层,进入有机相中。但这些针对界面的研究,主要集中在离子、分子水平。而在MSA体系中,可以通过显微成像手段直接地监测MSA在界面上的运动,从而更直观地分析获得萃取过程中界面上的作用力。
离子液体具有特殊的性质,其体系中存在复杂的超分子作用,对调控超分子组装行为具有很大意义。相比于传统有机溶剂,离子液体体系更易设计获得不同尺度的超分子组装体。目前已经报导的萃取体系中的MSA就是在离子液体体系中实现的。离子液体中阴阳离子种类繁多,可以对其结构进行各种设计。一方面可以设计特定的离子液体体系来获得所需的不同尺度的超分子组装,并基于此实现分离;另一方面还可以设计功能化的离子液体,使得形成MSA具有一些特殊的性质与功能,使MSA获得更广泛的应用。但是,如引言部分所述,未来的研究和应用中还需考虑提高传质速率和离子液体的损失与复用等问题。另外,离子液体辐照稳定性的研究主要集中在 γ辐照,在乏燃料后处理过程中α和β辐照效应将更为严重,但相关研究较少,有必要进一步深入。
猜你喜欢
军民两用技术与产品(2022年2期)2022-06-01
兵工学报(2022年2期)2022-05-22
兵工学报(2021年4期)2021-06-19
社会科学(2021年6期)2021-06-16
内蒙古民族大学学报(社会科学版)(2020年2期)2020-11-06
兵工学报(2020年12期)2020-02-06
科学导报(2018年30期)2018-05-14
电影故事(2016年19期)2016-11-10
太空探索(2016年5期)2016-07-12
科学中国人(2015年22期)2015-02-28
