
Efficacy and safety of Phillyrin (KD-1)capsule in the treatment of moderate COVlD-19:protocol for a randomized controlled trial
2022-03-08 08:16YangQingZhanRuiFengChenQinHaiMaJinPingZhengXiLongDengWeiYangLiFuNanShanZhongZiFengYang
TMR Modern Herbal Medicine 2022年1期
Yang-Qing Zhan,Rui-Feng Chen,Qin-Hai Ma,Jin-Ping Zheng,Xi-Long Deng,Wei Yang,Li Fu,Nan-Shan Zhong,Zi-Feng Yang,2*
1 National Clinical Research Center for Respiratory Disease,Guangzhou Institute of Respiratory Health,First Affiliated Hospital of Guangzhou Medical University,State Key Laboratory of Respiratory Disease(Guangzhou Medical University),Guangzhou,510230,China.2 Faculty of Chinese Medicine,Macau University of Science and Technology,Macau,9998078,China.3 Guangzhou Eighth People’s Hospital,Guangzhou,510060,China.
4 Guangdong Lewwin Pharmaceutical research institute Co.,Ltd;Guangzhou,510990,China.5 Jilin Yatai Chinese Medicine Development Institute,Changchun,130000,Jilin,China.
Abstract Objective Phillyrin (KD-1)is a traditional Chinese monomer and the main active component in Lianhua Qingwen.At present,sufficient studies have confirmed that KD-1 has significant anti-SARS-CoV-2 activity and antiinflammatory effects in vitro and in vivo.However,evidence-based studies to evaluate its therapeutic effect on COVID-19 are lacking.Therefore,we designed a clinical trial to evaluate the efficacy and safety of KD-1 in the treatment of moderate COVID-19 infection.Methods This is a multicenter,randomized,double-blind,placebo-controlled clinical trial.A total of 120 participants will be recruited and randomized to receive KD-1 capsule or placebo treatment for 14 days,50 mg per capsule,four capsules each time,three times a day.If the SARS-CoV-2 nucleic acid test results are negative twice within 14 days,the KD-1 capsule will be stopped the following day.Symptoms,patient compliance,and adverse reactions will be recorded,and nucleic acid testing will be conducted daily.Primary and secondary outcomes,as well as safety indicators,will be used to evaluate the efficacy and safety of theKD-1 capsule in the treatment of COVID-19.Discussion Herein,we describe the first clinical trial in China to treat COVID-19 using a traditional Chinese medicine monomer.A randomized,double-blind,placebo-controlled clinical trial is the best way to evaluate the efficacy and safety of KD-1 against moderate COVID-19 infection.If a good clinical benefit is observed,this represents the first step toward the use of KD-1 capsules to treat COVID-19.This clinical trial can serve as a model for other evidence-based research of traditional herbal medicines.Trialregistration This study is registered at chinadrugtrials.org.cn,with registration number:CTR20211800.
Keywords Phillyrin capsule,COVID-19,Pneumonia,Evidence-based clinical trial
Background
COVID-19,caused by the novel coronavirus SARS-CoV-2,has become an international public health emergency since its emergence in December 2019.By 29 June 2021,more than 180,000,000 COVID-19 cases have been confirmed,with over 3,900,000 deaths in 212 countries and regions.COVID-19 has caused considerable harm to public health and the economy.The emergence of multiple variants of SARS-CoV-2 has made the challenge of global prevention and control even more complicated.Despite the rapid spread of mutant strains,the typical symptoms of COVID-19 are still fever,dry cough,and fatigue.Less common symptoms include sputum production,headache,hemoptysis,diarrhea,anorexia,sore throat,chest pain,chills,and nausea,among other less common symptoms[1-4].In some cases,loss of smell or taste is the first symptom.Many infections are classified as mild[5,6];however,it is necessary to treat individuals with mild infection owing to the risks of developing severe illness and potential spread of the virus.
Traditional Chinese medicine is characteristic of Chinese medical science and an important part of health services.In the fight against COVID-19,Chinese medicine has played an active role in treatment,supported by strong evidence-based research[7-10].A multicenter,prospective,randomized controlled trial has shown that Lianhua Qingwen capsule can improve the rate of and reduce the time to symptom resolution,reduce the duration of fever,reduce fatigue and the severity of disease,shorten the length of hospital stay,and increase the cure rate[9].In vitro experiments have also confirmed that the Lianhua Qingwen capsule has an inhibitory effect on the replication of SARS-CoV-2,as well as cytokine disorders caused by infection with SARS-CoV-2[11].Those studies indicate that certain components of Lianhua Qingwen might provide a new resource for the development of anti-SARSCoV-2 drugs and have clinical benefits for the treatment of COVID-19 pneumonia.
Through literature research,we found that forsythia,as one of the sovereign drugs of Lianhua Qingwen,is one of the top ten traditional Chinese medicines in the 56 prescriptions for COVID-19 prevention in China[12],and also the first choice in the treatment of epidemic febrile disease[13].We guess forsythia plays an irreplaceable role in the prevention and treatment of COVID-19.Phillyrin(KD-1)is a quality control indicator ofForsythia suspensa(Thunb.)Vahl (regulated by the Chinese Pharmacopoeia).At the same time,the latest research has confirmed that KD-1 is one of the main active components of Lianhua Qingwen[7].So,we took the lead in vitro studies on the anti-COVID-19 activity of KD-1 and found that it could inhibit the replication of SARS-CoV-2 and inhibit cytokine disorders caused by the virus[14].Based on the evidence,we speculate that KD-1 is beneficial in the clinical treatment of COVID-19.
KD-1 has been purified fromForsythia suspensaby Dalian Fusheng Pharmaceutical Co.,Ltd.(Liaoning province,China),and developed into KD-1capsules with 90% active ingredient content.So,its quality and productivity have incomparable advantages over other medicinal ingredients.We think it has the potential to develop into a drug for the prevention and treatment of COVID-19.Therefore,we aim to perform a multicenter,prospective,double-blind,randomized controlled trial to verify the clinical efficacy oftheKD-1 capsule for the treatment of moderate COVID-19 infection.
Previous studies have suggested that KD-1 may inhibit SARS-CoV-2 in vivo and in vitro.Preclinical toxicology studies (including the acute,chronic,and special toxicological tests in different animals)and phase I clinical trials (ChiCTR-IIR-17011570)have shown that KD-1 is safe.Therefore,we aim to evaluate the efficacy and safety of the KD-1 capsule for the treatment of moderate COVID-19 infection in a randomized controlled trial.
Methods
The KD-1 capsule clinical trial will be conducted by the Declaration of Helsinki and Good Clinical Practice (GCP)guidelines.Ethical approval of this protocol was granted from the Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University (no.EC-2021-037(YW)-01;Guangzhou,China)as well as Guangzhou Eighth People’s Hospital (no,202110165;Guangzhou,China),and informed consent will be obtained from each participant before participating in the study.The main aim of this study is to investigate the efficacy and safety of KD-1 capsule in the treatment of moderate COVID-19 infection.To obtain accurate and objective conclusions,a randomized controlled trial will be performed.Febrile patients with moderate COVID-19 patients will be enrolled,according to preliminary study results that Lianhua Qingwen has a significant improvement effect on fever symptoms[9,15].
Study design
This clinical study is designed as a multicenter,randomized,double-blind,placebo-controlled trial in which eligible patients will be randomly assigned into two groups of equal proportions (1:1).One group will receive treatment with KD-1 capsule (treatment group)and the other group will receiveaplacebo.Based on current evidence for Lianhua Qingwen capsule in treating moderate COVID-19,patients will undergo 14 days of treatment and 21 days of observation.The flowchart of the clinical trial is shown in Figure 1.
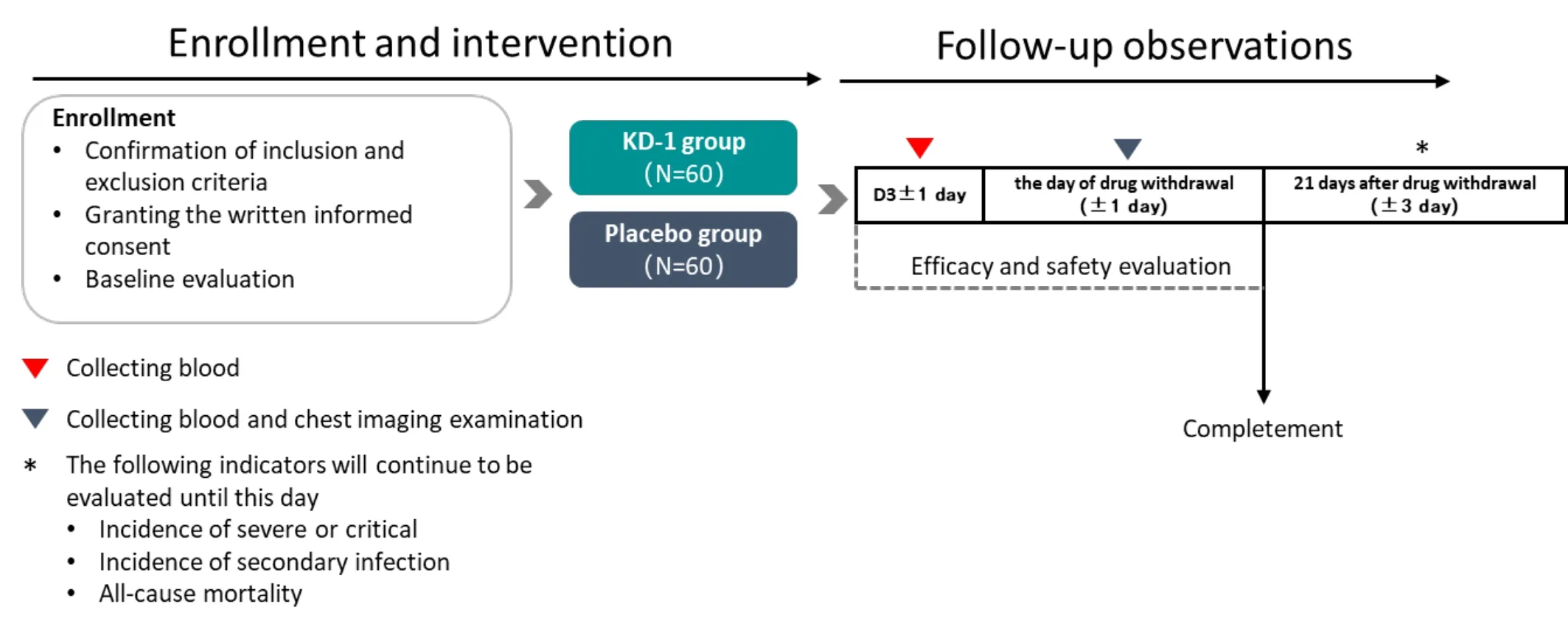
Figure 1.Study flowchart.
Staff training
To ensure patient safety,a blinded design,data quality,and compliance with the study protocol,personnel directly involved in the study all keep a GCP training certificate and will be retrained before the trial-.
Study population and participant enrollment
Participants in this trial will be recruited from the First Affiliated Hospital of Guangzhou Medical University,Guangzhou Eighth People’s Hospital,and other designated hospitals for COVID-19 treatment.Nasopharyngeal and throat swabs from patients will be tested to confirm SARSCoV-2 infection before enrollment.Patients meeting the inclusion criteria will be enrolled.If patients meet any of the exclusion criteria or termination standards,they will be removed from the trial.Finally,the included patients will be required to sign a written informed consent form,cooperate with doctors' treatment and follow-up,and provide information within the scope of ethical approval.Patients who do not fulfill these requirements will be removed from the study.
Inclusion and exclusion criteria
The inclusion criteria are patients (i)whose nasopharyngeal swab tested positive for SARS-CoV-2 virus nucleic acid by real-time reverse transcriptionpolymerase chain reaction (RT-PCR),(ii)aged 18 to 65 years,(iii)with axillary temperature≥37.5°C (within 24 hours of onset and a course≤48 hours,or fever appearing 24 hours after onset and a course≤72 hours;course start time is the time of onset of primary clinical symptoms of COVID-19 including axillary temperature≥37.5°C,or cough score≥2 points,or fatigue score≥2 points),(iv)with chest imaging showing pneumonia,(v)who agree to voluntary participation in the study and sign the written informed consent.The exclusion criteria are detailed in Table 1.
Rejection criteria or termination standards
The rejection criteria are (i)not meeting the inclusion or exclusion criteria,(ii)withdrawal of informed consent,(iii)not cooperating with doctors' treatment and follow-up after selection,(iv)any serious breach in the procedure(incorrect administration leading to effects that cannot be judged,e.g.,receiving other therapies simultaneously such that efficacy cannot be determined).Termination standards refer to patients who meet the inclusion criteria but who must be terminated during the trial and who are included in the final statistical analyses.The study may be suspended or prematurely terminated for sufficient and reasonable causes.The party that terminates the trial should submit a written notice and record the reason for termination to the participants,researchers,and clinical testing units including the pharmaceutical supervisory and administrative departments.Termination standards are detailed in Table 2.
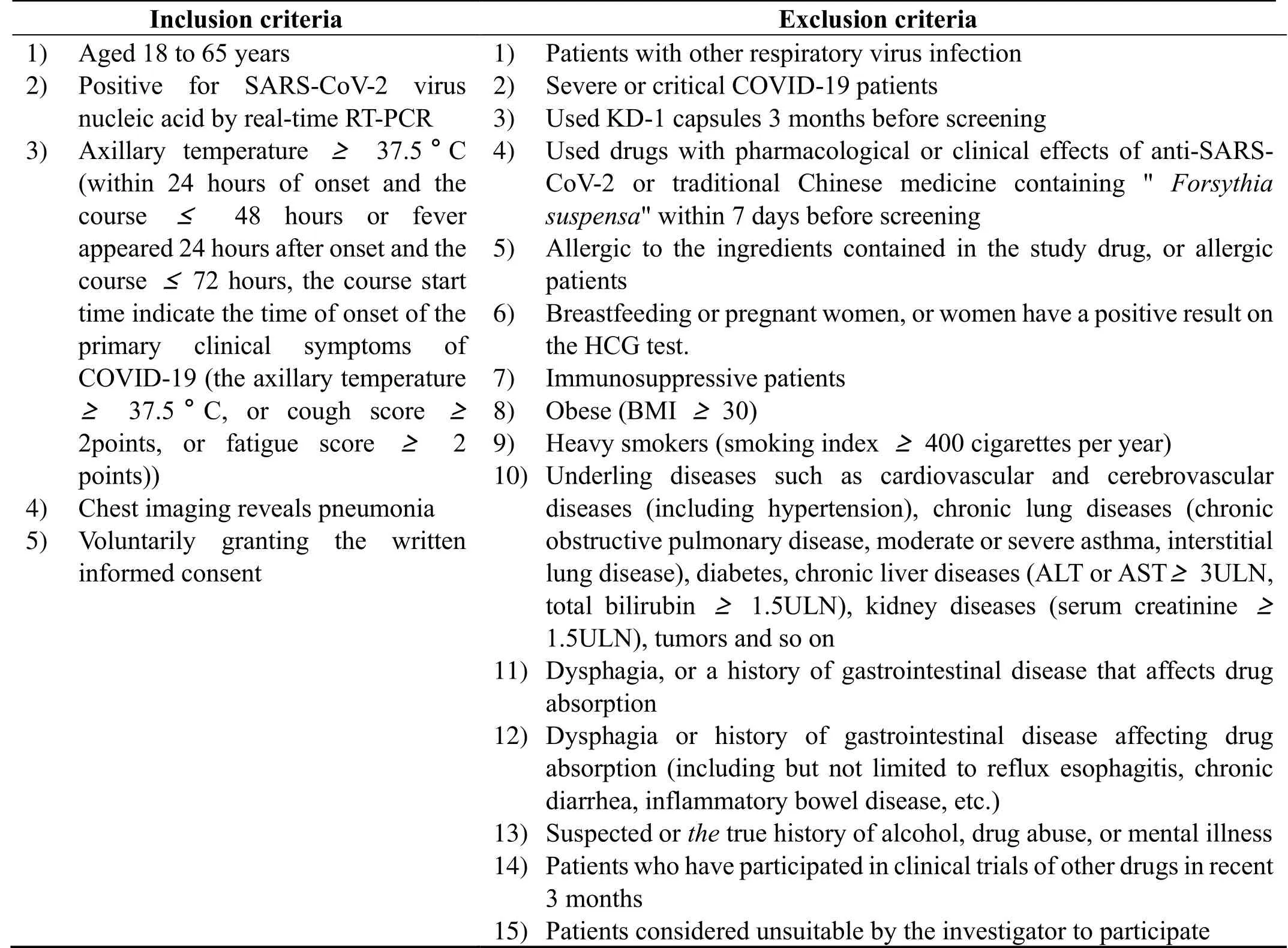
Table 1.Inclusion and exclusion criteria
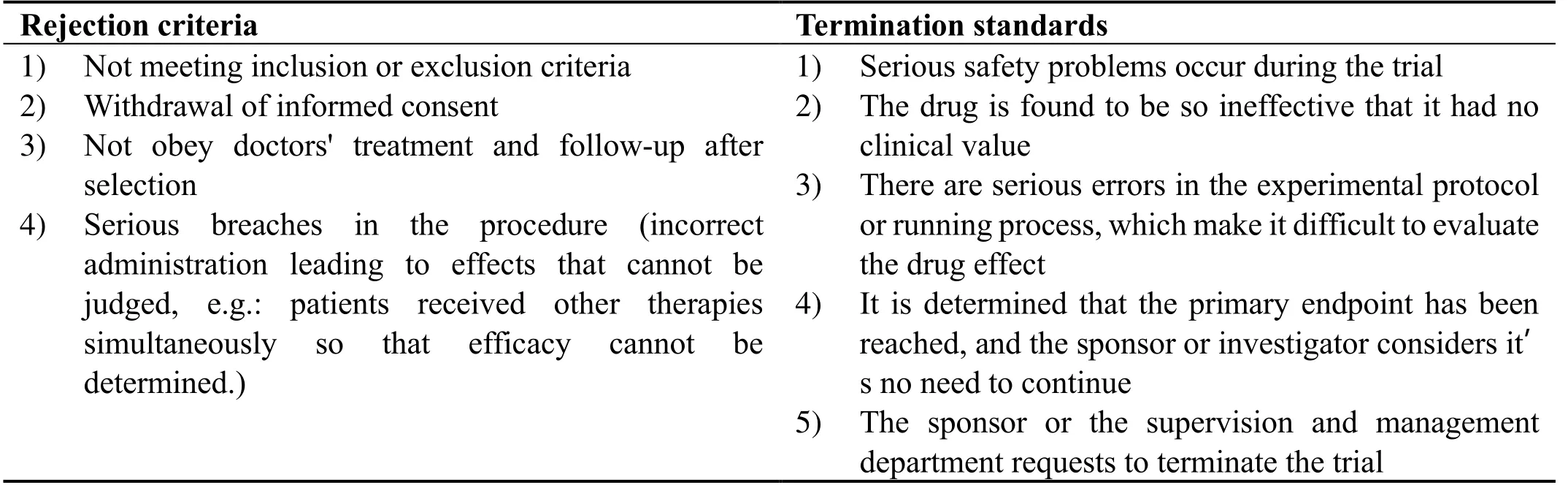
Table 2.Rejection criteria or termination standards
Intervention
KD-1capsuletreatmentgroupFour KD-1 capsules (50 mg/per capsule)will be taken orally per time,three times daily,for 14 days.
PlacebogroupFour placebo capsules (50 mg/per capsule)will be taken orally per time,three times daily,for 14 days.Both KD-1 capsules and placebo are produced and packa-ged by Dalian Fusheng Pharmaceutical Co.,Ltd.(Liaoning province,China).The placebo only contains the same excipients (corn starch),matches the KD-1 capsules in taste and color,and follows the same dosage and directions for treatment as for KD-1 capsules.According to the requirements of a double-blinded test,all drugs used in this double-blinded trial are counted and sorted by a third party(Chinese Evidence-based Medicine Center,China).All drugs are within the period of validity.
If a participant’s body temperature returns to normal within 14 days,respiratory symptoms are significantly improved,and SARS-CoV-2 nucleic acid test results are negative twice with an interval longer than 24 hours,the participant can stop taking KD-1 capsule or placebo on the following day.If the nucleic acid test result is still positive 14 days later,the patients should continue treatment according to the COVID-19 Diagnosis and Treatment Protocol[16].Follow-up will continue for 21 days after the cessation oftheKD-1 capsule.
GeneraltreatmentAccording to ‘Zero COVID-19 Case Policy’,all people entering China will be required to conduct health screening such as body temperature detection and nucleic acid detection,and at the same time,they will be required to conduct 14 days of home or intensive medical observation.Once the nucleic acid test is positive,people will be transferred to designated hospitals for medical observation or treatment immediately in the closed-loop.Regular nucleic acid monitoring will be carried out for staff with high exposure risk.Once the nucleic acid screening is positive,health observation measures will be put into place for that closely connected and sub-closely connected personnel.
As for treatment,referring to the current version of the COVID-19 Diagnosis and Treatment Protocol,all participants will be allowed to receive the best possible supportive care,mainly including:(a)bed rest and supportive treatment to ensure adequate energy intake,with special attention to water-electrolyte balance to maintain a stable internal environment,and close monitoring of vital signs and oxygen saturation;(b)routine blood examination,urine dry chemistry analysis,C reactive protein,biochemical indexes (liver enzymes,myocardial enzymes,renal function),coagulation function,arterial blood gas analysis,and chest imaging monitored according to the participant’s condition;(c)effective oxygen therapy provided promptly.
BanneddrugsandtreatmentsBecause there is currently no clear and effective drug treatment for COVID-19,no positive drug group was set up in this trial.To ensure the efficacy and safety of KD-1 capsules in treating COVID-19 and avoid the effect of drug interference and drug interaction,the following provisions are made for banned drugs and/or treatments.
(1)Traditional Chinese medicines (including decoction,Chinese patent medicine,Chinese injection,and Chinese medicated diet)recommended for the treatment of COVID-19 in the current COVID-19 Diagnosis and Treatment Protocol should not be used during the trial.
(2)Drugs with pharmacological or clinical effects against SARS-CoV-2 should not be used during the trial.
(3)Acupuncture,cupping,moxibustion,and other traditional Chinese medicine physiotherapy measures should not be used during the trial.
Emergency medicine and ethics
Investigators will decide whether to use emergency treatment if a participant has axillary temperature≥38.5°C for more than 4 hours or has a body temperature>39°C.If neither of these situations occurs,then acetaminophen tablets are preferred for fever.If a participant has a cough score≥2 points,the investigators will decide whether to issue drugs,such as compound methoxamine capsules.All emergency drugs will be provided by Dalian Fusheng Pharmaceutical Co.,Ltd.If the patient's condition worsens further,investigators will treat the patient according to the actual situation and decide whether to terminate their participation in the experiment.
All participants will be informed in detail about the purpose,methods,and process of the study and be provided with satisfactory answers to any queries before their formal enrollment.All enrolled participants will agree to voluntary participation in the study and will be informed that they can withdraw from the study at any time with no unfavorable consequences.
Data collection and outcome measures
DatacollectionandmanagementElectronic Data
Capture (EDC)will be used in this study.The investigator completes the source documents (including but not limited to outpatient and inpatient records,laboratory examination reports,participant medication diary cards,and specific items collected for each follow-up period,as outlined in Table 3).An authorized clinical research coordinator will organize the study records and assist the investigators in entering the data into the EDC system.If there is any question regarding the data,the data manager will query the investigator or other authorized person,who must respond to the data query as soon as possible.Based on the response,the manager will confirm the data and may reissue a data query if necessary.Information on medical history,adverse events,and combination therapy collected in clinical trials will be coded using standard dictionaries;medical coding should be done before locking the data.After a blinding review and confirmation of corrections in the established database,the data will be locked by the principal investigator,sponsor,statistical analyst,and data manager.Once locked,the data are generally not allowed to be unlocked.If the data need to be unlocked,the unlocking conditions and process must follow the corresponding standard operating procedures,and the unlocking process must be carefully controlled and recorded.The locked data will be assessed by confirming the intent-to-treat set (ITTS),full-analysis set (FAS),perprotocol set (PPS),and safety set (SS).The decision will be approved by the data manager,sponsors,and investigators and will end with unblinding after locking the database.
Outcome measures
Primary outcomeThe primary outcome is the remission rate of major clinical symptoms(abbreviated as RR in the calculation formula)within 14 days,which is defined as body temperature (axillary temperature)≤37°C,cough,and fatigue score≤1 point,and maintenance of stable symptoms for 24 hours.The following is the calculationformula.And the scoring standard of clinical symptoms is shown in Table 4.
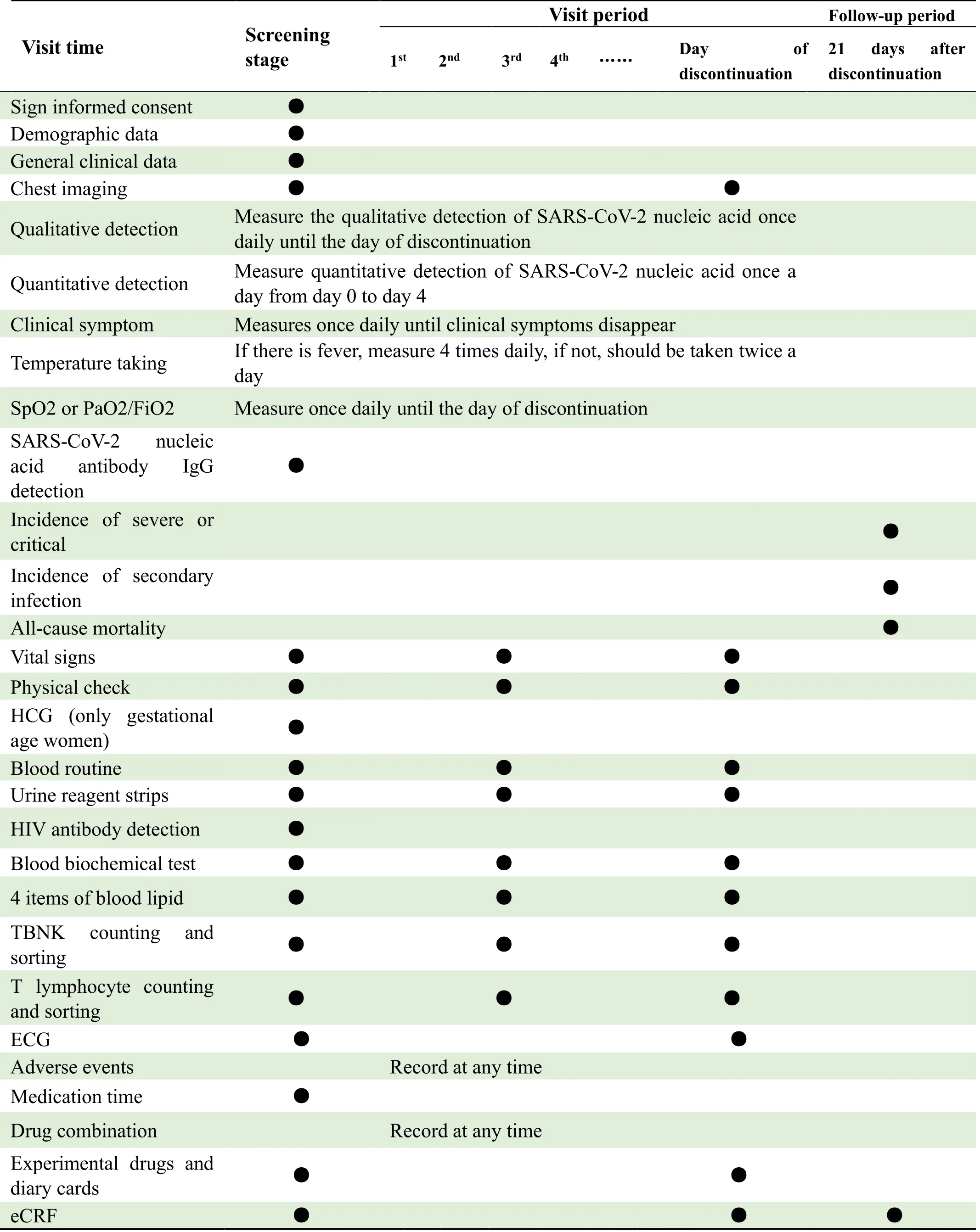
Table 3.Chart of treatment using KD-1 capsule against moderate COVID-19 infection

Table 4.Scoring standard of clinical symptoms
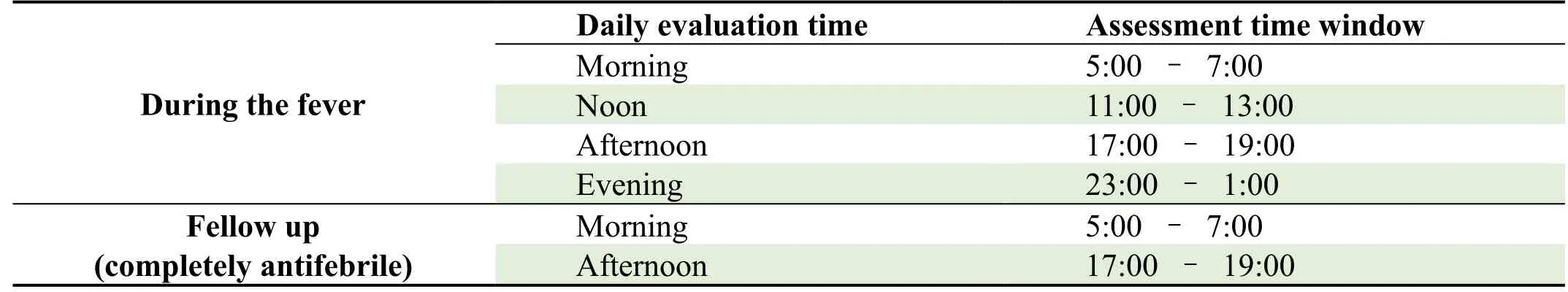
Table 5.Temperature-monitoring schedule

Secondary outcomesThe secondary endpoints consist of the following:
(i)Duration of fever.Body temperature normalization means axillary temperature≤37°C,with maintenance for more than 24 hours.To accurately assess fever recovery time,temperature measurement should be carried out in strict accordance with the schedule (Table 5).
(ii)Recurrence rate of fever after normalization.
(iii)Clinical remission rate(abbreviated as CRR in the calculation formula)within 14 days.This endpoint is defined as body temperature (axillary temperature)≤37°C,with maintenance of normothermia for more than 3 days(inclusive),respiratory symptoms score≤1 point (cough,sore throat,myalgia,and others),and significant improvement in acute exudative lesions.The following is the calculation formula:

(iv)Rate and time of virus nucleic acid negative conversion within 14 days.The standard of negative conversion is as follows:nucleic acid testing of airway samples is negative in two consecutive tests (at least 24 hours between samples),and the time to virus nucleic acid negative conversion is the time to first negative conversion.
(v)Remission time of major clinical symptoms.
(vi)Variation in SARS-CoV-2 viral load in nasal or pharyngeal swabs on days 1–4.
(vii)Incidence of severe and critical cases during the trial.
(viii)Incidence of secondary infection from the first day of receiving medication to day 21 after stopping medication.
(ix)All-cause mortality from the first day of receiving medication to day 21 after stopping medication.
Safety evaluationSafety assessment is another important purpose of this trial.Vital signs,physical examination,electrocardiography,and clinical laboratory tests(including routine blood tests,blood and urine lipids,liver and kidney function,myocardial enzymes)will be tested during the screening inspection period,on day 3,and the day following the cessation of medication.Additionally,adverse reactions will be observed and recorded at any time during the trial.
Evaluation of adverse eventsAn adverse event refers to all adverse medical events that occur after the participant receives the test drug,which may manifest as symptoms and signs,diseases,or abnormal laboratory examination results but that may not have a causal relationship with the test drug.According to theCommonTerminologyCriteriaforAdverse Events(CTCAE)v5.0,adverse events are divided into five levels (see Table 6).Serious adverse events occur when the agent:
(i)results in death;
(ii)is life-threatening;
(iii)requires hospitalization or extends the length of hospital stay;
(iv)causes permanent or severe disability/incapacity;
(v)leads to congenital abnormalities and birth defects;
(vi)leads to other important medical events (such as cancer).
All serious adverse events should be reported to the State Food and Drug Administration and the ethics committee within 24 hours after the researchers learn of an event.The causal relationship between adverse events and the test drug will be determined using the standards shown in Table 7.
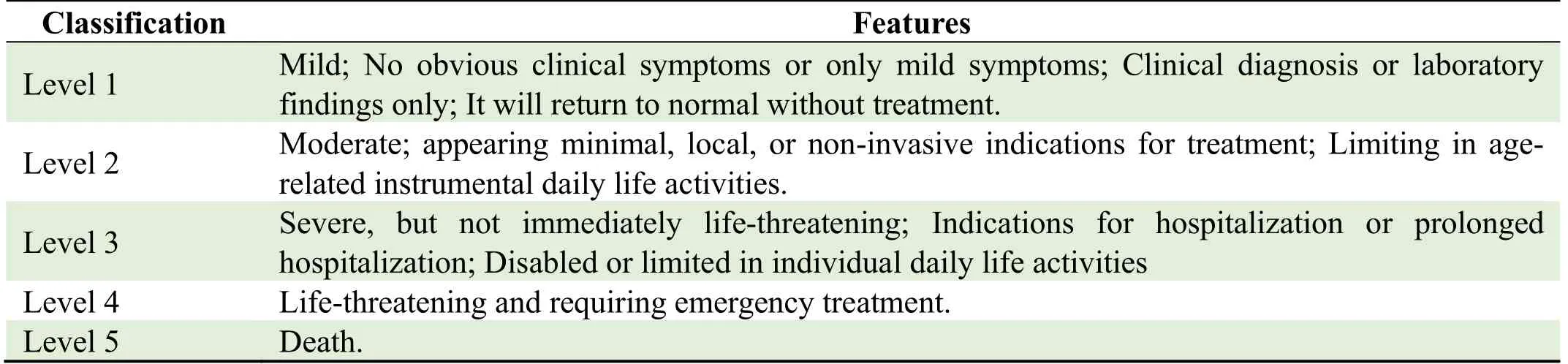
Table 6.Classification of disease severity
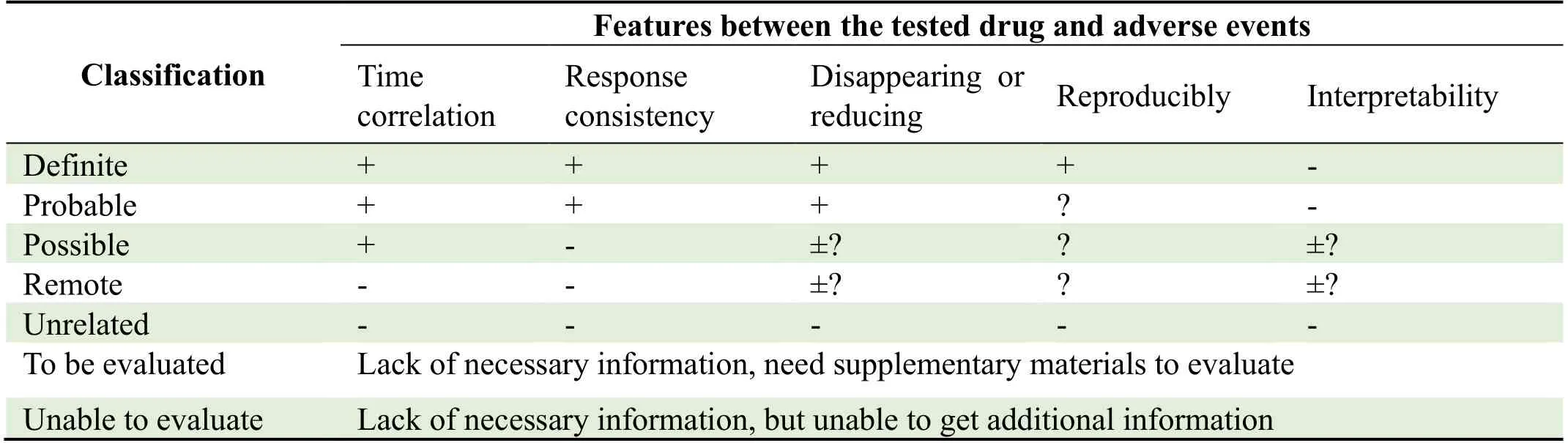
Table 7.Classification of adverse events
Statistical analysis
Sample size calculationThe primary outcome of this trial is the remission rate of major COVID-19 symptoms within 14 days.Assuming that the remission rate of major symptoms on day 14 is 65% in the placebo group and 90%in the treatment group,the sample size will be calculated using the Group Sequential Proportions module in PASS,taking α=0.025 (one-sided)and power=80%.Thus,52 participants per group will be required.considering the dropout rate;the final study population will include 120 participants with 60 patients per group.Because estimation of the sample size in this trial lacks clinical evidence,after half of the participants complete evaluation of the primary outcome,an interim analysis will be performed by the Independent Data Monitoring Committee (IDMC).The sample size required for this study may be recalculated and adjusted based on the results of the interim analysis.
Patient groups for data analysesIn this study,patients will be classified into four categories:ITTS,FAS,PPS,and SS.The ITTS will include all participants who provide their informed consent and accept randomization.The FAS will comprise participants who receive treatment at least once after randomization and for whom corresponding curative effect index records can be obtained.The PPS is a subset of the FAS,including all participants who complete drug treatment according to the protocol,with no majorprotocol deviation,and complete all of the evaluation content.The standards for the PPS population will be finalized during the blinding review,including at least the following conditions:
(i)meets the selection criteria;
(ii)completed all planned visits;
(iii)no use of drugs or treatments that may affect the evaluation of a curative effect during the trial;
(iv)having good compliance (80%–120%).SS refers to participants with safety index records.
In this trial,the ITTS will be used to perform the baseline data and efficacy analysis,The PPS will also be used to analyze the primary efficacy,but the main conclusions will be based on analysis of the ITTS.When the analysis results are consistent between the ITTS and PPS,the credibility of the trial conclusions will be enhanced.For patients in the FAS who fail to adhere to the specified treatment process,the last observation carried forward (LOCF)will be used.The SAP will mainly be used to estimate the laboratory data and adverse events.The total participants in the SS will be taken as the denominator of the incidence of adverse events.
Statistical analyses(i)Statistical software:SAS version 9.4 or above statistical software will be used for statistical analysis (SAS Institute,Cary,NC,USA).
(ii)Basic principles:All statistical inferences should be carried out in two-sided tests.P<0.05 will be set to indicate statistical significance,and two-sided 95% confidence intervals (CIs)will be used
(iii)Missing data:The LOCF method will be used when a participant’s record has incomplete information during the trial.
(iv)Extreme data:Sensitivity analysis should be used when important indicators involve extreme data.
(v)Abscission analysis:Inclusion and completion will be summarized,as well as the list of abscission cases and the number of participants in the FAS,PPS and SS.We will use a flow chart to describe the distribution of participants and list the causes of participant loss at each point in each group.
(vi)Descriptive statistics:Measurement data will be reported as mean,standard deviation (SD),and CIs,and the minimum,maximum,P25,median,and P75 will be given when necessary.The mean,SD,and CIs of differences will be applied to paired measurement data.Enumeration and ranked data shall be reported using the number of participants and the corresponding percentage.
(vii)Primary endpoint:The Clopper–Pearson method will be used to calculate the 95% CI as well as the rate difference between the two groups and its 95% CI.The Cochran–Mantel–Haenszel chi-square test or accurate probability method will be used for statistical tests.
(viii)Secondary endpoints:Parts of secondary endpoints (specifically,the clinical remission rate,virus nucleic acid negative conversion rate,symptom resolution rate)will use the same statistical methods as the primary endpoint.Some secondary endpoints (like the time to body temperature recovery,time to symptom relief,time to symptom resolution)will use Kaplan–Meier plots;the median time and its 95% CI will be calculated using the Greenwood method.Log-rank tests will be used for difference analysis,and a Cox proportional hazards model will be applied to estimate the hazard ratio and its 95% CI.Changes in viral load will be analyzed in covariance analysis,using the participant's baseline viral load as a covariable.
(ix)Safety analysis:The Pearson’s χ2 test or Fisher’s exact test will be used to compare the incidence of adverse events between groups,and adverse events will also be listed and described.Normal and abnormal laboratory results will be recorded,as well as the relationship between changes and test drugs.
(x)Baseline data:According to the distribution of variables,quantitative data will be analyzed using analysis of variance (ANOVA)or a nonparametric statistical method.Qualitative data will be analyzed using the χ2 test or Fisher’s exact test,and the Wilcoxon rank-sum test will be used for ranked data.
ClinicalmonitoringDuring the experiment,clinical units must comply with GCP and the clinical trial scheme.Pharmaceutical supervisory and administrative departments will arbitrarily conduct audit inspections and will cooperate with the sponsor to assign medical doctors.We will ensure that participants will receive appropriate treatment for adverse events during the trial.Any moderate or serious adverse events will be reported to the Ethics Committee and the pharmaceutical supervisory and administrative departments.The sponsor shall bear the cost of treatment and corresponding economic compensation in case of injury or death related to the trial.
Discussion
There is still no specific medicine available to treat COVID-19.Treatment for COVID-19 continues to be focused on symptomatic treatment as well as the treatment and prevention of complications such as acute respiratory failure.The development of drugs against SARS-CoV-2 is of critical importance while the COVID-19 pandemic continues globally and the virus continues to mutate.Our trial aims to be the first clinical trial to use a monomer of Chinese herbs to treat cases of moderate COVID-19 infection in China.According to the results of this pilot study,we will conduct a large-scale clinical trial to fully evaluate the efficacy and safety oftheKD-1 capsule against COVID-19.It is hoped that the KD-1 capsule for the treatment of moderate COVID-19 infection will be fully verified.
As one of the main active ingredients of Lianhua Qingwen,we assume that the therapeutic effect oftheKD-1 capsule on COVID-19 will have similar characteristics.We will select patients with moderate COVID-19 infection for study inclusion,according to previous clinical studies of Lianhua Qingwen in the treatment of COVID-19[9,15,17,18].In that study,Lianhua Qingwen was found to significantly improve the resolution rate of major clinical symptoms (fever,fatigue,and cough),reaching 57.7%after 7-day treatment,80.3% after 10-day treatment,and 91.5% after 14-day treatment[9].Therefore,in this study,the primary outcome indicator is set to be the remission rate of major clinical symptoms on day 14.Studies have shown that among clinical symptoms,Lianhua Qingwen significantly improves fever symptoms,with a remission rate of 95.77%[19].More importantly,theKD-1 capsule has already obtained the approval of the State Food and Drug Administration for fever symptoms caused by influenza viruses (No.2016L10731).Therefore,secondary indicators of this trial will focus on fever symptoms.
In hospitalized patients,the median duration of measured fever is 7 days (interquartile range 3–11 days)and the median duration of subjective fever is 8 days(interquartile range 4–13 days)[20],indicating that the duration of fever symptoms in some patients with COVID-19 is short.Fever is the most common symptom of COVID-19[1,5,21,22] and an important efficacy evaluation index in our trial.To ensure statistical efficiency and achieve our study aims,we will have strict criteria for patient inclusion.These patients must have developed fever within 24 hours of onset with a course≤48 hours,or fever must appear 24 hours after onset with a course≤72 hours.
The higher viral load may increase the risk of death in hospitalized patients[23,24].For the treatment of COVID-19,drugs with potential anti-SARS-CoV-2 effects should be used early[25].Although no specific drugs against SARS-CoV-2 are available at present,some drugs may have certain therapeutic effects in clinical observation.Studies have shown that convalescent plasma therapy can reduce SARS-CoV-2 viral load,thereby reducing the risk of dying from COVID-19[26].REGN-COV2,a neutralizing antibody cocktail,reduces viral load and results in a significantly lower percentage of COVID-19-related medical visits,suggesting that virus clearance is associated with better clinical outcomes[27].Therefore,this trial is of great value in terms of determining whether KD-1 can improve the rate of and shorten the time to virus nucleic acid negative conversion as well as reduce the incidence of severe or critical illness.
The key characteristics in assessing the quality of a drug are its safety and efficacy.We designed this trial as a multicenter,randomized,double-blind,placebo-controlled,pilot study to evaluate the safety and efficacy of KD-1.We hope that the trial results will provide evidence in support of KD-1 capsules to treat moderate COVID-19 and prevent the development of severe illness.
Data availability
Correspondence and requests for materials should be addressed to jeffyah@163.com(Prof.Zi-Feng Yang).
Abbreviations
COVID-19,2019 novel coronavirus infection;RT-PCR,reverse transcription-polymerase chain reaction;HCG,human chorionic gonadotropin;ULN,the upper limit of normal;BMI,body mass index;ALT,alanine aminotransferase;AST,aspartate transaminase;SpO2,oxygen saturation;PaO2,arterial oxygen partial pressure;FiO2,fractional inspired oxygen;ECG,electrocardiogram;Ig,immunoglobulin;TBNK,T lymphocyte subsets,B lymphocyte subsets,and NK cells;eCRF,electronic case report form;IDMC,the Independent Data Monitoring Committee.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (No.82174053).This research received a grant from Dalian Fusheng Pharmaceutical Co.,Ltd.(Liaoning province,China).Thanks to all medical staff and investigators for their efforts in this outbreak.Thanks to Shanghai Renzhi Data Technology Co.,Ltd.for donating the central randomization system and providing technical support for this study.We thank Dalian Fusheng Pharmaceutical Co.,Ltd.for providing the drugs to the patients in this study for free.Thanks to Guangzhou Evidence-based Medicine Tech Co.,Ltd.for assisting in the planning and implementation of this study.
Author contributions
Yang-Qing Zhan and Rui-Feng Chen contributed equally to the work;they revised this protocol.Nan-Shan Zhong and Zi-Feng Yang conceived, designed,and coordinated this study.Qin-Hai Ma,Jinping Zheng,and Xi-Long Deng participated in the implementation of the project and ethical approval;Wei Yang provided statistical advice.Li Fu sought funding.All authors contributed to the writing of the manuscript and read and approved the final manuscript.
Competing interests
The authors declare no competing interests.Fortunately,this trial was funded by Dalian Fusheng Pharmaceutical Co.,Ltd.
 TMR Modern Herbal Medicine2022年1期
TMR Modern Herbal Medicine2022年1期
- TMR Modern Herbal Medicine的其它文章
- lndirubin suppresses 4T1 murine breast cancer in vitro and in vivo by induction of ferroptosis
- Speed-resolved blood perfusion and oxygen saturation in human skin response to thermal stimulation
- Clinical practice guidelines for traditional Chinese medicine and integrated traditional Chinese and western medicine:a cross-sectional study of data analysis from 2010 to 2020
- Guizhi Fuling capsule can induce apoptosis of myeloma cells through the mitochondrial apoptosis pathway