
无序碳单层的电子结构及氢原子吸附的第一性原理研究
2022-03-04 02:32刘佳溪孙志海夏永鹏邹勇进张焕芝黄鹏儒孙立贤
原子与分子物理学报 2022年4期
张 颖,刘佳溪,孙志海,黄 强,夏永鹏,李 彬,邹勇进,张焕芝,黄鹏儒,徐 芬,孙立贤
( 桂林电子科技大学材料科学与工程学院 广西信息材料重点实验室,桂林 541004;广西新能源材料构效关系协同创新中心,桂林 541004)
1 引 言
二维材料的探索与研究,能够促进基础科学的发展.二维材料因为其独特的结构及优异的物理、化学特性,在多个领域具有潜在的技术应用价值.其中,石墨烯是一种蜂窝状晶格排列的零带隙半金属二维材料,通过微机械剥离方法成功地从石墨中分离出来,这促使了研究人员对具有独特性能的新型2D 材料的研究[1-3].然而,在石墨烯的生长或加工过程中,完美的蜂窝晶格往往会出现结构缺陷.例如Stone -Wales 缺陷[4],此晶体缺陷涉及到两个π 键连接的碳原子的变化,即连接两个碳原子的键围绕键的中点旋转90 度,可将四个六边形转换成两个五边形和两个七边形.
尽管缺陷的存在破坏了石墨烯晶格完美的对称性,并影响了其某些性质,如降低载流子的迁移率[5,6],但缺陷对其他性质的影响是有益的.例如,缺陷可以增加石墨烯的反应活性,并可用于调整其在催化应用中的电子性能.因此,将非六边形缺陷引入六边形晶格是重建碳原子、改善电子性质的有效方法.目前,探索具有独特晶体结构、机械和电子性能的新型二维碳的同素异形体的工作层出不穷[7-36].所提出的结构是由包含不同边数的多边形构成的,如4 + 8 环[7],5环[8],4 +6 +8 环[9,10,20-22],4 +5 +6 +8 +10环[11],5 +7 环[12-14],和5 +6 +7 环[13,15-19].然而,这些理论上提出的具有改进性质的二维碳的同素异形体多用于研究其力学性质和电学性质.因此,设计和探究新型的催化类二维碳的同素异形体具有重要的意义和价值.
表面吸附可以调节材料的电子结构,从而达到改变其表面性能的目的.特别是对于二维材料体系来说,它们拥有比其他材料较大的比表面积,可以通过吸附外来原子提高二维材料体系的化学活性.例如: 氢原子吸附于石墨烯可改变其电子和磁性结构.此外,氢原子与石墨化合物的相互作用在核聚变[37,38],氢储存[39]等许多领域也有着重要作用.
在这项工作中,我们采用第一性原理计算方法研究了无序碳单层(5 +6 +8 +11 环) 的晶体结构、电子结构及其对氢原子吸附的性质,进而研究其催化性能.探讨无序性导致的不同配位及局域结构对C 原子键长、原子电荷、电子分布的影响.通过在不同点位上进行氢原子吸附探讨C 原子位点分布及化学活性.该研究将在一定程度上揭示二维无序碳材料结构-性能的构效关系,为实验上设计新型催化类的无序碳功能材料提供理论指导.
2 模型构建与计算方法
基于密度泛函理论,本研究采用VASP 程序包进行计算[40-42].投影缀加波( PAW)[43,44]方法,用于处理电子-离子之间的相互作用,在改进的广义梯度近似( GGA) 下,使用Perdew - Burke -Ernzerhof( PBE) 泛函形式来处理交换关联函数.由于GGA 和局部密度近似LDA[45]不能准确的计算由于波动电荷分布之间的动力学相关性引起的范德华相互作用,本研究添加色散修正DFT -D3的方法来减小误差[46,47].根据石墨烯固有缺陷的类型构建二维无序碳结构的初始结构.无序碳为全碳分子的二维平面网状,晶格参数为a=b=17.22 Å,真空层c=10 Å.结构优化过程平面波的截断能为500 eV,k-point 设置为6 ×6 ×1.体系的能量收敛标准为1 ×10-5eV.
为了研究了材料表面氢原子的吸附性能.无序碳吸附氢原子的吸附能定义如下:
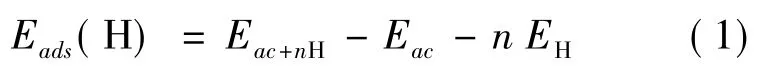
其中Eac+nH吸附H 原子后体系包含的总能量,Eac是无序碳的总能量,n 为吸附H 原子的数量,EH为自由的H 分子中H 原子的总能量.吸附能越负,对应的H 分子与H 原子吸附越强,模型越稳定.
对于给定模型的析氢性能( HER) 与单个氢原子的吸附能密切相关,同时吉布斯自由能越接近0,材料的析氢性能越优越.因此我们计算了吉布斯自由能,公式如下:
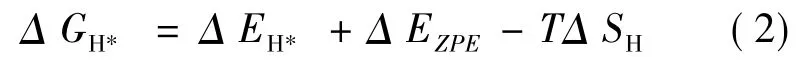
其中Δ EH*是吸附氢原子的能量,Δ EZPE是零点吸附能的变化.对于Δ SH,一般取Δ SH≈-1/2( Δ SH2) ,其中Δ SH2是气相氢分子在标准条件下的熵.因此公式(2) 可以重新写为[48]:
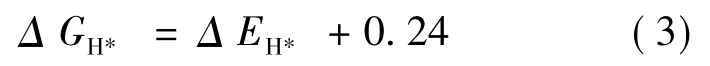
其中0.24 eV 是298 K 时ZPE 和熵的组合对表面模型的贡献.通常认为吸附状态下的氢吸附的振动熵可以忽略不计.
3 结果与讨论
3.1 单层无序碳的几何结构
图1 为基于石墨烯的固有缺陷调节得到的无序碳结构.经优化体系达到稳定后,体系的形成能为34.475 eV,较大的形成能是因为体系内存在较多的断键或较强的内部应力.石墨烯的碳碳键长为0.142 nm[48],但该体系中的碳碳键长与石墨烯中的键长有明显不同,键长在0.13 -0.18 nm 之间,与石墨烯的碳碳键长相比最大的变化范围达到26.7%.如图1 所示,C-C 键被拉长的有C6- C20,C7- C22,C15- C29,C23- C35,C36-C48,C46- C47,C40- C41,C53- C68,C55- C69,C67-C80,C69-C84; 被压缩的有C17-C18,C40-C41.因此,多元环的引入会导致附近碳原子的键长发生不同程度的拉伸或压缩.原子间键长的变化会引起电化学性能的改变[48].为了研究无序多元环的引入对体系电子结构的影响,进一步计算了体系的电子态密度( DOS) 以及电子局域密度函数( ELF).
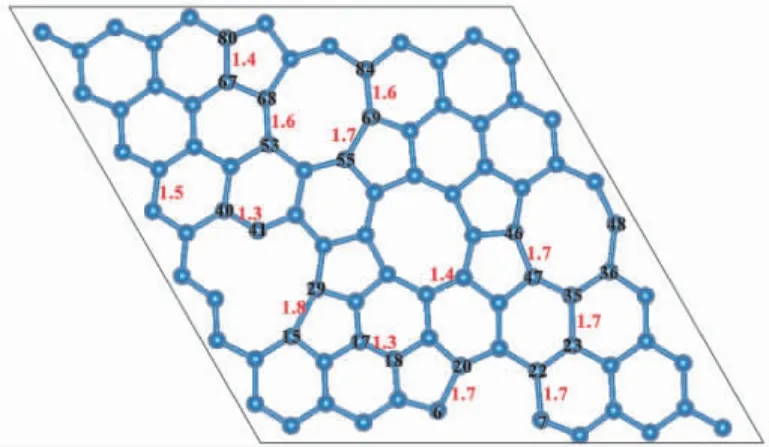
图1 无序碳单层晶体结构Fig.1 Crystal structure of amorphous carbon monolayer
3.2 单层无序碳电子结构
在完美的六角晶格中,石墨烯在费米能级处形成狄拉克锥电子结构,电子有效质量小,迁移率大,使其呈现出类金属的性质[49].六角晶格的破坏改变了这一电子结构,如图2( a) 所示,在费米能级附近的锥形结构消失,出现较多局域电子峰,体系的电子态增加.这些电子峰的出现使得部分C 原子的电子局域性增强,化学活性发生改变.有研究报道,析氢活性起源于电子结构的变化,结构内电子态的改变可以为析氢反应提供丰富的活性位点[50].为此,本研究计算了单层无序碳的电子局域函数,考察不同位点处C 原子附近电子的局域特性及化学键性质的改变,如图2( b)所示,可以看到,体系中电子主要集中在两个C原子之间,例如C17- C18,C40- C41原子键长较短,约0.13 nm,其碳原子间的ELF 值≈0.95 表现出极强的共价键性质; 部分碳碳键长被拉伸处于0.16 ~0.18 nm 区间内( 如: C6- C20,C7-C22,C15- C29,C23- C35,C36- C48,C46- C47,C53-C68,C55-C69,C67-C80,C69-C84) ,其碳原子间的ELF 值相约为0.8 ~0.85.同时,在出现断键又无法形成C -C 键的原子处会出现孤对电子,例如: 图中C28原子只形成2 个配位,悬挂键为未配对电子,具有极强的局域性.C28的局域结构极不稳定,很容易通过吸附外来原子进行电子配对从而降低形成能.
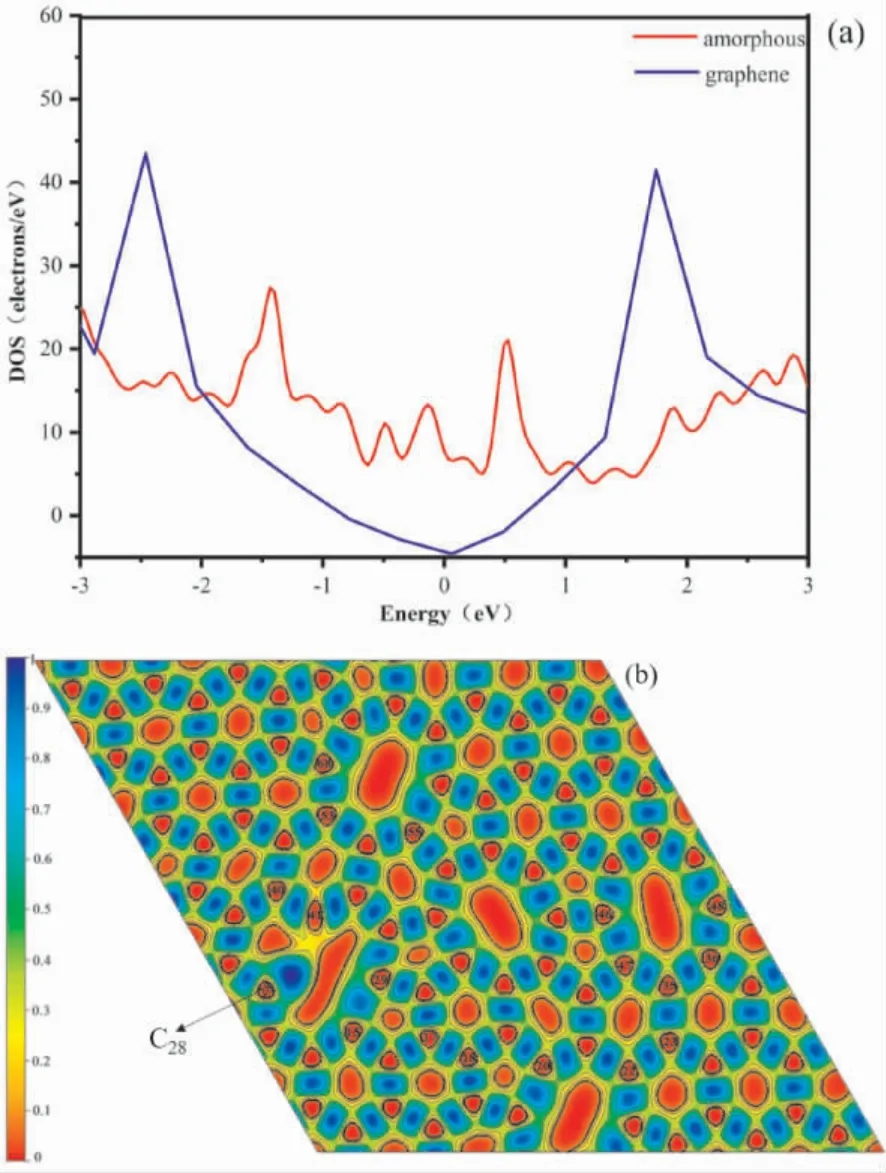
图2 ( a) 无序碳单层与石墨烯的电子密度对比和( b) 电子局域密度函数Fig.2 ( a) Comparison of electronic density of states between amorphous carbonmonolayer and graphene; ( b) electronic local density function
图3( a) 为单层无序碳结构的二维电荷密度分布情况.与图2( b) 相比可以看出电荷密度分布与原子键长、电子局域性相对应.电荷密度相对越高,电子局域密度越大.体系中电荷聚集在C15-C16,C17-C18,C18- C19,C29- C30,C40- C41键长较短处,键长较长的碳碳键( C6- C20,C7-C22,C36-C48,C46-C47,C55-C69,C69-C84) 几乎没有电荷的聚集.不同键长的碳原子改变了体系的电子结构,增加了体系的聚集电荷和局域电子,使得不同碳原子位点可能表现出不同的电荷强弱.在电荷密度的基础上对每个碳原子的Bader电荷进行了计算,Bader 电荷布居[51]是根据电荷零通量面分割的,通常认为该方法能够较好的定义分配到原子上的电子数[52-54].图3( b) Bader 电荷可以看出无序碳中的电子较多的原子为C20,C41,C67,失电子较多的原子的为C18,C21,C32.因为几何结构变化的多样性,结构内电子转移呈现多样性.图3( c) 和( d) 分别是费米面下方和费米面上方区间的电荷分布图.从图中我们可以看出,最高占据态主要由C40,C41,C15,C14,C21,C22,C9等原子贡献,最低未占据态主要由C15,C32,C21,C34,C52,C53等原子贡献.从图3( c)和( d) 中可看出,提供电子占据态的原子均位于多元环与六元环的交界位点.综上所述,由于体系中局部六角晶格被破坏,无序性增加,改变了体系电子结构造成部分原子局域性增强.
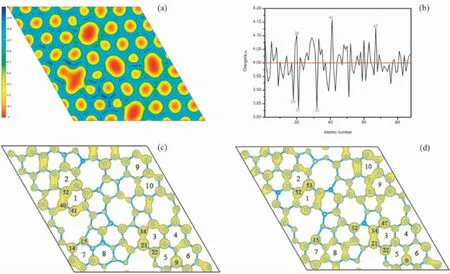
图3 单层无序碳( a) 电荷分布情况; ( b) C 原子电荷布居; ( c) 费米面下方附近[-0.05,0]eV 区间的电荷密度; ( d) 费米面上方[0,0.05]eV 区间的电荷密度Fig.3 Amorphous carbon monolayer: ( a) charge distribution; ( b) C atom charge distribution; ( c) charge density in the[-0.05,0]eV interval near the bottom of the Fermi surface; ( d) above the Fermi surface[0 ,0.05]charge density in the eV interval
3.3 单层无序碳的氢吸附性能
上文中系统地分析了无序碳结构中的晶体结构和电子性质,发现C6,C20,C69等原子位点的碳碳键长较短,电荷聚集,电子局域,通常认为这些位点的化学活性较高.C15,C20,C40,C53,C68等原子位点碳碳键长较长,电子局域性较差,通常认为化学活性较低.接下来对这些位点的吸附性能进行定量的计算和研究.通过在相应的碳原子上方添加一个氢原子进行结构优化,如图4所示为优化后得到的氢原子吸附结构图,可以看出对于大部分的吸附点,氢原子吸附于碳原子的上方属于顶位吸附,这主要是氢原子与碳原子的pz轨 道 成 键.例 如: C17,C20,C27,C32,C53,C68,C69.同时,一些位点的C -H 键会与体系处于同一平面,主要是因为氢原子可能与碳原子的sp2杂化轨道成键.可以看出吸附后C -H 键长与标准碳氢键( 0.109 nm)[55]相比变化范围在0.0001 nm ~0.0028 nm 之间.吸附后氢原子位于无序碳平面内结构的C -H 键长较垂直于无序碳平面的C-H 键长变化较小,进一步说明氢原子占据sp2轨道形成的C-H 键键强较强.
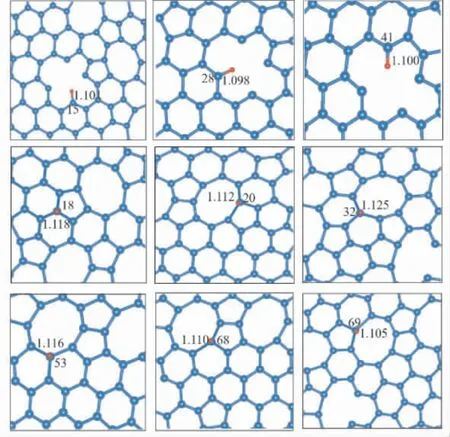
图4 单层无序碳氢原子吸附的俯视图模型Fig.4 Hydrogen adsorption model of amorphous carbon monolayer
图5 为吸附前后态密度对比图.不同位点氢原子吸附后总态密度峰的位置发生变化,在费米能级附近H( s) 与C( p) 轨道发生大部分的重合与少量的杂化.吸附后体系的总态密度较吸附前产生较大的差异,以C15为例费米能级附近的局域电子峰减少.C15,C28两位点氢原子的态密度曲线均有一个较高的峰值,氢原子轨道占据较高; 其他位点如C17,C20等吸附后氢原子的态密度较平坦,离域较大.氢原子的成键方式以及轨道的占据情况会对其吸附能造成一定的影响,如图6( a)C15,C28两个位点的吸附能分别为-1.76 eV, -2.21 eV 较其他吸附位点吸附能低,吸附后结构更稳定.单个原子的吸附能与解离能的强弱是析氢反应发生的关键[56-60]图5、图6( a) 以及表1的数据综合分析,不同位点不同键长以及键角对氢原子的吸附能力不同.例: C15,C28,C40三个位点呈现出二配位结构,对氢原子的吸附能较大,但其过大的吸附能导致氢原子的解离相对较难;C68,C69二者在多元环结构中所处位置的键角较大,导致其吸附能较大; C17,C18两原子位于五元环与六圆环之间且键角较平均,对氢原子的吸附能力较弱.综上,通过调节共价键的性质可以调整其成键方式,不同的成键方式会影响其吸附氢原子的能量.氢原子的成键方式不同,导致各位点的吸附能以及解离能不同,从而为析氢反应产生更多的活性位点.
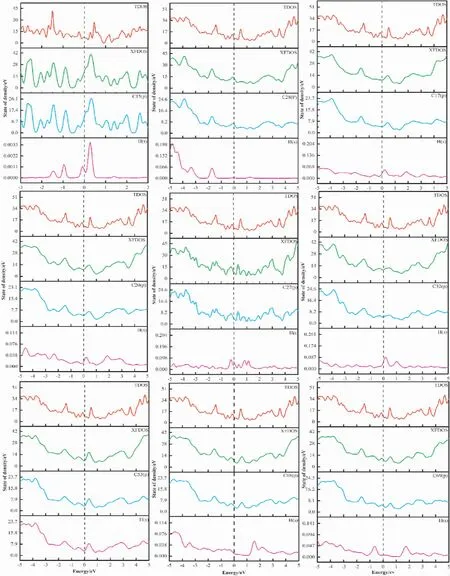
图5 吸附前的总态密度与各位点吸附后总态密度、各吸附碳原子以及氢原子态密度的对比图Fig.5 Comparison diagrams of the total density of states before adsorption and that after adsorption on each site,the densities of states of each adsorbed carbon atom and hydrogen atom.
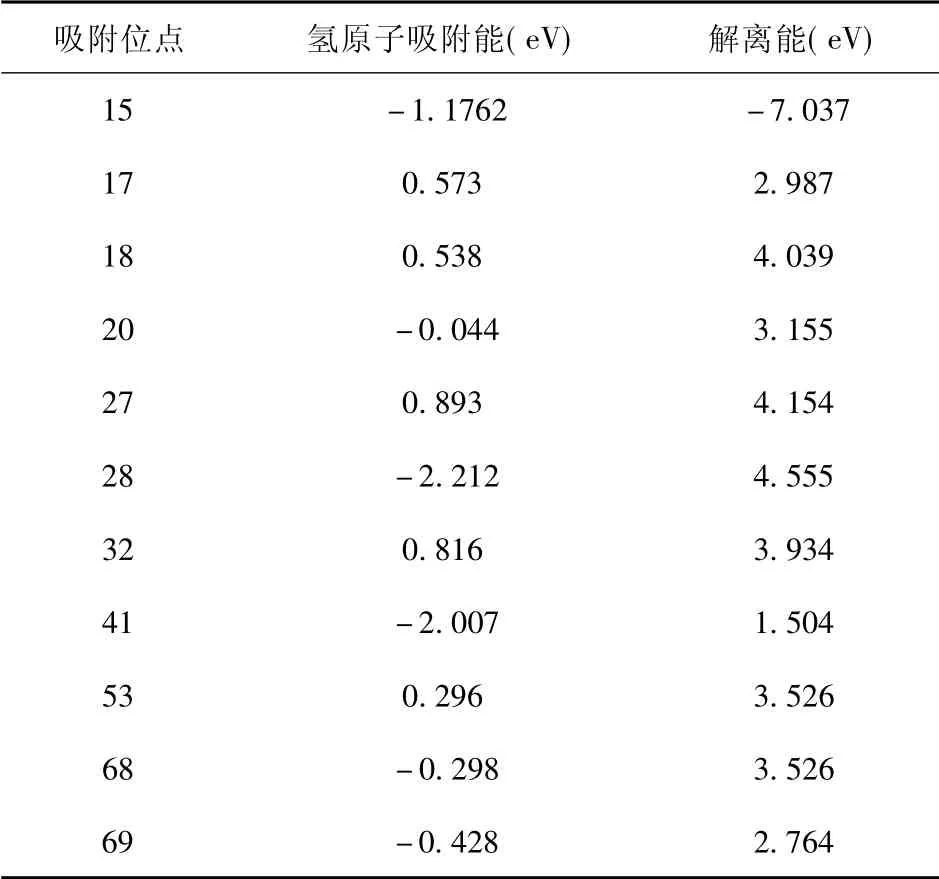
表1 不同位点氢原子解离能Table 1 Dissociation energies of hydrogen atom at differentsites
为了更好地探索无序碳材料析氢反应的性能,计算了不同位点对氢吸附的吉布斯自由能如图6( b).对于良好的析氢反应催化剂,如Pt 的ΔGH*值接近于零,其代表着对于氢原子的吸附和释放强度适中[58].图6( b) 中C53,C68位点的吉布斯自由能与Pt 的吉布斯自由能接近.两位点均为三配位具有较大的键角且pz轨道与氢原子成键.因此无序碳材料可以通过自身电子结构变化引起活性位点的增加为析氢反应提供良好的催化剂.
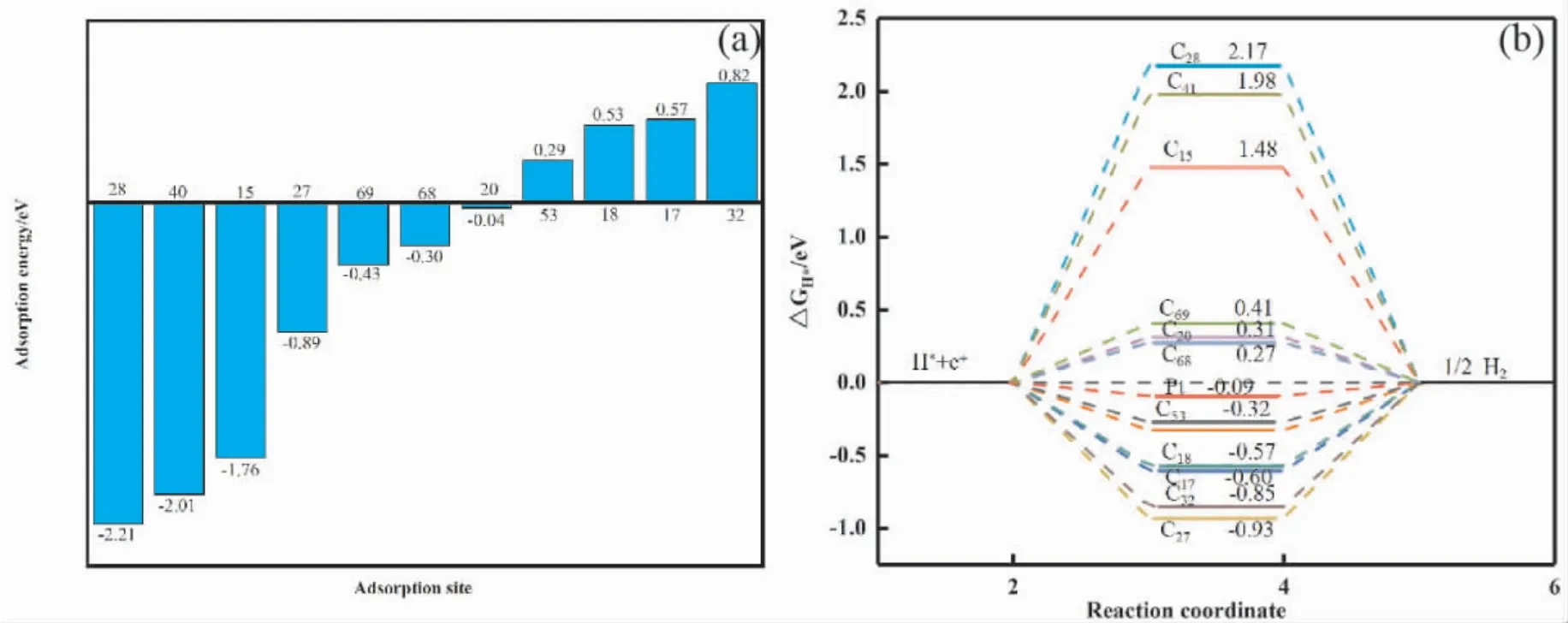
图6 无序碳单层( a) 不同吸附位点的吸附能和( b) 无序碳吸附氢原子的吉布斯自由能Fig.6 Amorphous carbon monolayer ( a) adsorption energies of different adsorption sites and ( b) Gibbs free energies
4 结 论
本研究运用基于密度泛函理论第一性原理计算研究了二维单层无序材料的几何结构、电子结构及其析氢性能,得到如下结论:
1) 体系中缺陷的引入,改变了部分位点的键长、电荷密度以及电荷转移量和电子局域性,使材料中增加了大量的性能各异的吸附位点,例如:C53,C68,C15,C28等.
2) 通过对不同位点晶体结构和吸附性能的对比,发现C53,C68吸附氢适中具有较好催化潜力.这些位点的特征是与相邻碳原子具有三个配位,并且三个sp2共价键的对称性被打破,其中一个键角增大.
3) 通过调控在六元环与多元环交界处构造三配位且键角较大的位点可以调节吸附氢性能使其具有较好的析氢性能.
猜你喜欢
舰船科学技术(2022年21期)2022-12-12
物理学报(2022年11期)2022-06-18
山西大学学报(自然科学版)(2021年5期)2021-12-25
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中国科技纵横(2018年3期)2018-03-15
中学物理·高中(2016年8期)2016-08-08
枣庄学院学报(2015年5期)2016-01-09
中国医学装备(2015年10期)2015-12-29
中学化学(2015年8期)2015-12-29
火炸药学报(2012年4期)2012-01-29
