
A novel SPINT2 missense mutation causes syndromic congenital sodium diarrhea
2022-03-01 05:45:06XiaXuZhangXiChenWeiZhouVasilisCaesarMavratsasYangYangXiaoXinRuiTanSongJiaZhengXingXingZhang
World Journal of Pediatrics 2022年12期
Xia n-Xu Zhang · Xi Chen · Wei Zhou · Vasilis Caesar Mavratsas · Yang-Yang Xiao · Xin-Rui Tan · Song-Jia Zheng · Xing-Xing Zhang
Congenital sodium diarrhea (CSD) is a monogenic disorder caused by specific genetic defects that increase sodium content in the stool, resulting in intractable diarrhea. The disease was first reported by Holmberg in 1985 [ 1]. Fewer than 50 cases have been reported worldwide to date, mainly in Europe (Germany and Sweden) and Oceania (Australia), while no cases have been reported in China. There are two categories of CSD depending on whether it involves other congenital malformations: non-syndromic congenital sodium diarrhea (non-sCSD) and syndromic congenital sodium diarrhea (sCSD). For non-sCSD, the identified causative genes includeSLC9A3andGUCY2C, whereas sCSD is primarily caused bySPINT2mutations [ 2].
SPINT2, also known as hepatocyte growth factor activator or inhibitor type 2 (HAI-2), is expressed in most epithelial cells. It is a membrane-bound, serine protease inhibitor containing two Kunitz structural domains (KDs): Kunitz structural domain 1 (KD1) and Kunitz structural domain 2 (KD2) [ 3]. In humans,SPINT2is generally considered to play a crucial role in organogenesis and intestinal ion homeostasis [ 4]. The types ofSPINT2gene mutations mainly include missense, nonsense and frameshift mutations, with one of the most common mutations being c.488A > G (p.Tyr163Cys) [ 5]. However, the relationship betweenSPINT2gene mutations and the onset of sCSD remains poorly understood.
CSD is clinically and genetically heterogeneous, often leading to missed diagnoses and misdiagnoses; so further study of this condition is needed. In the present study, we reported a case of sCSD in China’s Hunan Province, which carried a previously unreported homozygous mutation (c.386A > G, p.Y129C) in theSPINT2gene that we believed could be a novel pathogenic mutation based on clinical symptoms and family co-segregation analysis. Rare clinical cases of mutations require further detailed studies, as emphasized in their publication by Iwanieczyk et al. [ 6]. Analyzing the clinical features and genetic alterations in this case of sCSD will contribute to the understanding and management of this rare disease.
The proband was the first child of healthy parents without consanguinity. Routine prenatal screenings were conducted during the mother’s pregnancy. There was no prenatal history of infection, exposure to radioactive substances, polyhydramnios or fetal anomalies (Fig. 1 a). This child was born at full term with a birth weight of 3.53 kg, a normal height of 50 cm, and a normal Apgar score. On the day he was born, the patient started having four to five yellow–green paste-like stools (without pus, blood, or mucus) per day (Fig. 1 d). He was exclusively breastfed but without weight gain during the first 17 days. The child got a high fever (peaking at 39.6 ℃) accompanied by abdominal distension, more frequent diarrhea (8–12 episodes of yellow–green watery stools without acidic odor per day), severe dehydration and shortness of breath, resulting in 0.65 kg of weight loss. Inpatient treatment led to the resolution of the child’s dehydration and infection, and his weight slowly increased to 3.65 kg, but diarrhea did not improve. In the following three months, the patient was referred to several hospitals, but his diarrhea persisted, and his bodyweight gradually decreased. Upon admission to our hospital, he was 108 days old with a bodyweight of 3 kg and height of 52 cm, showing a retardation of growth. The child was extremely thin, with inelastic skin and almost no subcutaneous fat. Physical examination revealed a distended abdomen with active bowel sounds and an enlarged liver. He did not exhibit anal atresia, cleft palate, or posterior nasal atresia (Fig. 1 c). Laboratory screening revealed hyponatremia at 126 mmol/L (130–150 mmol/L), hyperkalemia at 5.8 mmol/L (3.5–5.5 mmol/L), stool sodium of 121 mmol/L and hypoproteinemia at 28.3 g/L (40–55 g/L). Computed tomography (CT) of the abdomen showed obvious gas and fl uid accumulation in the intestines (Fig. 1 e). Colitis and proctitis were revealed by colonoscopy, but no colorectal deformities were observed. A biopsy of the pathological tissue was performed resulting in normal acid-fast staining and revealing only moderate chronic colitis, and there was no evidence of active infl ammation or enterocyte vacuolation. During the course of treatment, there were a series of complications, the most common of which were infections of the respiratory, urinary and digestive tracts, which resulted in exacerbation of the patient’s diarrhea (Fig. 1 f). At one year of age, the patient developed a combination of secondary hemophagocytic lymphohistiocytosis (sHLH) due to prolonged intravenous nutrition and uncontrollable multisystemic infections according to hemophagocytic lymphohistiocytosis (HLH) 2004 criteria (Supplementary Table 1) [ 7]. Unfortunately, he died of multiple organ failure at the age of 17 months despite having undergone five months of conventional chemotherapy (methylprednisolone, cyclosporine and etoposide).
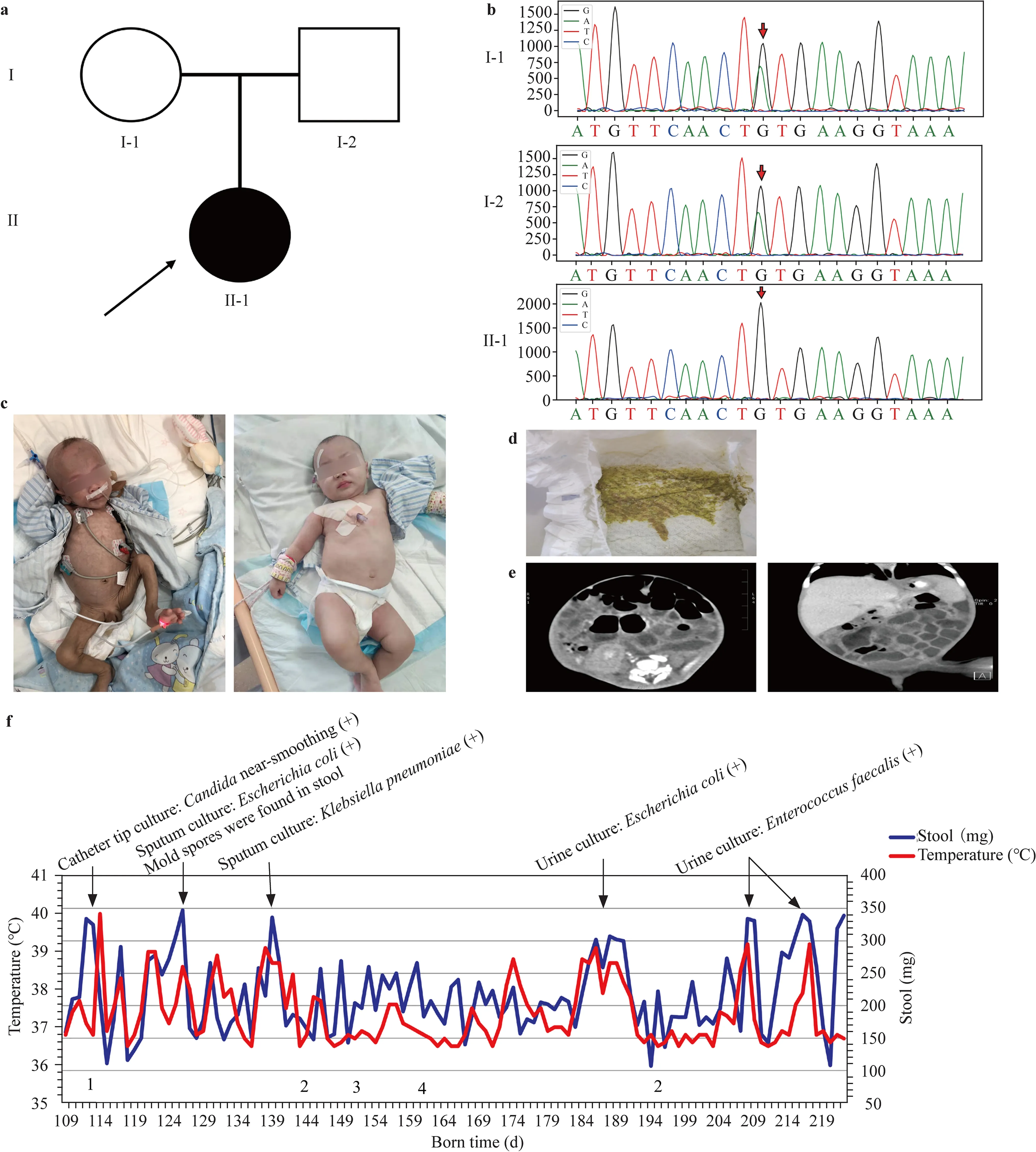
Fig. 1 a Pedigree of the family showing a segregating autosomal recessive form of sCSD. Clear circles and squares represent normal females and males, respectively. The black symbol represents the affected individual (proband). b Sanger sequencing of the patient and his parents. c Left: clinical manifestations at three months of age. Right: clinical manifestations at 10 months of age. After six months of treatment, the patient improved significantly. d Stool characteristics: yellow–green watery stool. e Abdominal CT at four months of age: fl uid and gas accumulation in the intestinal canal, mild dilatation and slight thickening of the small intestinal wall (transverse section on the left, coronal section on the right). f 1: Venous catheter thrombosis, 2: hepatic impairment, 3: BCG-associated lymphadenitis, and 4: pathological fracture of the humerus. During hospitalization, the child had intermittent fever complicated by multiple infections, including infections of the respiratory, urinary, and intestinal tracts as well as infections caused by intravenous access; diarrhea tended to worsen significantly in the presence of infection. sCSD syndromic congenital sodium diarrhea, CT Computed tomography, BCG Bacille Calmette–Guérin
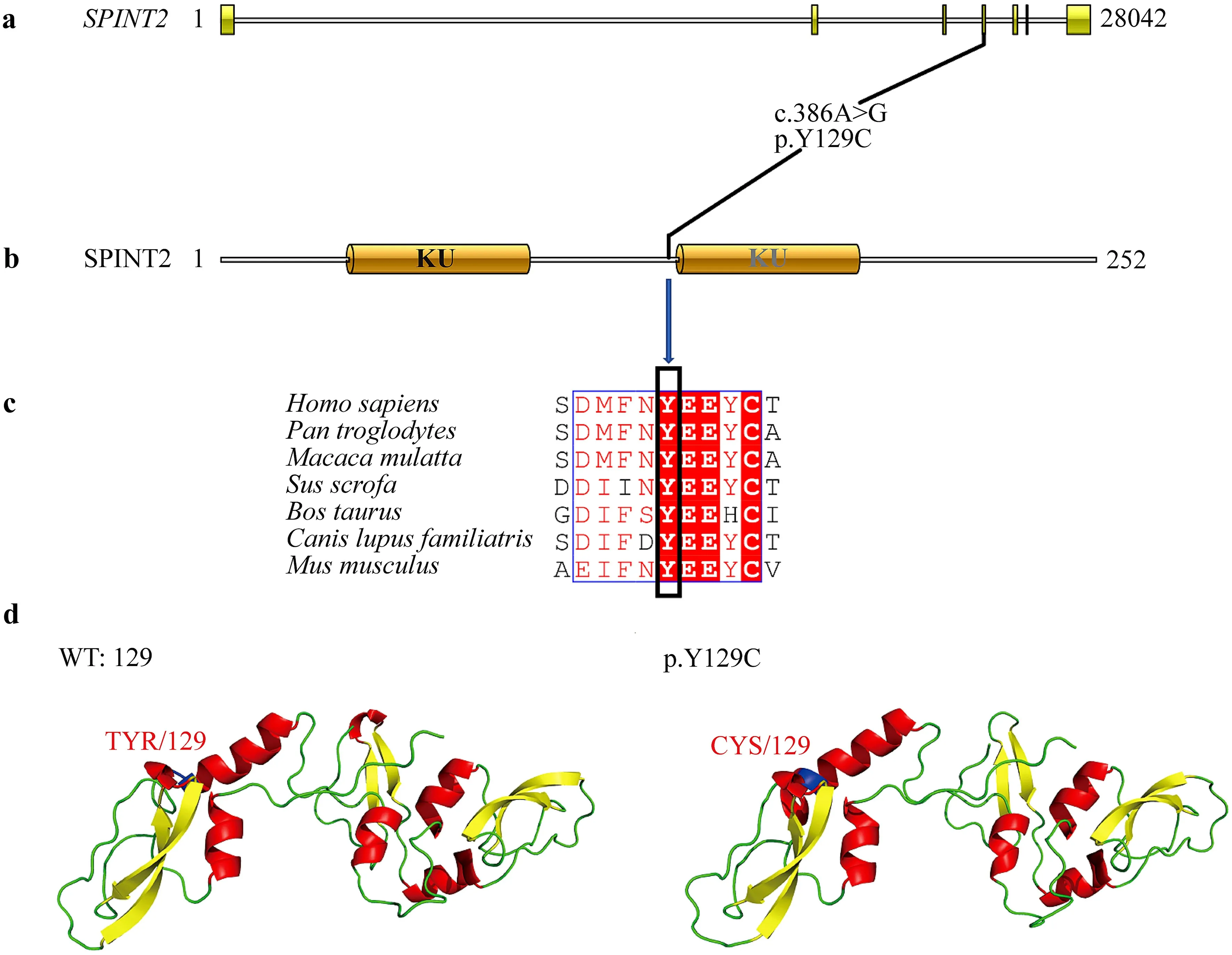
Fig. 2 The SPINT2 amino acid 129 is a conserved region and predicted protein structure. a SPINT2 gene structure, where the yellow color indicates the exonic region of the gene. The black line indicates information about the position of the mutation site on the gene. b SPINT2 protein conserved structural domain prediction. SPINT2 contains two Kunitz structural domains (KDs): Kunitz structural domain 1 (KD1) and Kunitz structural domain 2 (KD2). c Conservation analysis of a variety of SPINT2 amino acids at position 129 by multiple sequence alignment (ClustalW). Organisms aligned include Homo sapiens, Pan troglodytes, Bos taurus, Macaca mulatta, Sus scrofa, Bos taurus, Canis lupus familiaris, Rattus norvegicus and Mus musculus. d Left is WT: 129 wild-type SPINT2 protein structure; the blue region is the tyrosine at position 129. Right is p.Y129C mutant protein structure; the blue regionis the cysteine at position 129. WT wild type
Intractable diarrhea with failure to thrive and exclusion of infectious diseases guided us in performing trio exome sequencing and one exon of theSPINT2gene was sequenced by Sanger sequencing. Genomic DNA was extracted with the magnetic bead method, using a blood genomic DNA extraction kit (Tiangen Biotech, Beijing, China). Qualified genomic DNA samples were recovered by fragmentation processing using a Covaris M220 Focused-ultrasonicator and were processed in preparation for whole human gene exome sequencing using the xGen®Exome Research Panel v2.0 kit of Integrated DNA Technologies (IDT, USA). Liquid phase capture wasused to build a library. The captured DNA library was used to perform 2 × 150 bp paired-end sequencing of the human whole gene exome sequence through the Illumina HiseqX sequencing platform (Illumina Company, USA). Finally, ANNOVAR ( http:// annov ar. openb ioinf ormat ics. org ) software was used to perform mutation site point annotations and to obtain candidate pathogenic mutation sites. A homozygous missense mutation (c.386A > G; p.Y129C) in the fourth exon of theSPINT2gene was detected and verified by Sanger sequencing (Fig. 1 b). The more conserved an amino acid is during species evolution, the more important this amino acid is for maintaining the function of the protein. A multiple sequence alignment of SPINT2 amino acid sequences revealed that amino acid site 129 is conserved in common species, such asHomo sapiens,Pan troglodytes,Bos taurusandMus musculus(Fig. 2).

Table 1 Clinical and molecular findings of sCSD according to different SPINT2 sequence variants based on literature review
Based on structural analysis of the SPINT2 mutant protein, we further analyzed the mutation at this locus using mutation deleteriousness prediction software including SIFT, Polyphen2 and MutationTaster (Supplementary Table 2). Most software predictions indicated that the mutation p.Y129C negatively affected protein function.
TheSPINT2gene contains two conserved serine protease inhibitor Kunitz domains, which are rich in disulfide bonds formed between cysteine residues. Although our mutation is not in the Kunitz structural domain, the mutation of tyrosine to cysteine at position 129 will likely affect disulfide bond formation in the Kunitz structural domain. In this study, modeling of wild-type and mutant proteins was performed using YASARA software (Fig. 2 d). Our results demonstrate that the conformation of the β-fold within the KD2 structural domain changed after the tyrosine at position 129 mutated to cysteine, which increased the length of the α-helix in front of the KD2 structural domain. On the whole, the p.Y129C mutation causes the spatial conformation near the KD2 structural domain of the SPINT2 protein to become tighter than the wild type, which could affect the binding ability of the SPINT2 protein.
The most important clinical features and pathological laboratory values seen in sCSD patients according to the type ofSPINT2sequence variant are compiled in Table 1. The following complications were reported in individual patients: hemophagocytic lymphohistiocytosis, chronic intestinal inflammation, central line infections, gastrointestinal motility disorder and osteoporosis.
In currently published case reports, no deaths have been found in non-sCSD patients and 13 deaths have been reported in sCSD patients [ 4, 8]. Two of these deaths were caused by recurrent catheter-related infections, and five deaths were due to complications relating to intravenous nutrition. These results demonstrated that in addition to the severe dehydration and disturbance of the internal environment caused by the disease itself, the complications of sCSD also present high-risk factors for death. Although an in-depth and comprehensive assessment of sCSD is not possible owing to the limited number of reported cases, early diagnosis and timely treatment can reduce mortality and can improve the prognosis.
Supplementary InformationThe online version contains supplementary material available at https:// doi. org/ 10. 1007/ s12519- 022- 00613-6.
Author contributionsThe first ZXX: conceptualization, data curation, methodology, writing–original draft, writing–review and editing. CX: writing–original draft, writing–review and editing. ZW: writing–original draft, writing–review and editing. MVC: writing–review and editing. XYY: data curation, methodology, writing–review and editing. TXR: data curation, methodology, writing–review and editing. ZSJ: data curation, methodology, writing–review and editing. The second ZXX: writing–review and editing.
FundingThis research was funded by the Natural Science Foundation of Hunan Province (2021JJ0137).
Data availabilityThe data that support the findings of this study openly available in Science Data Bank https:// doi. org/ 10. 11922/ scien cedb. 01571 (unregistered). Data private access link https:// www. scidb. cn/s/ baM3Ab .
Declarations
Conflict of interestNo financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.
Ethical approvalThis study has been approved by Ethical Committee of Second Xiangya Hospital of Central South University (approval number: K052), and informed consent has been obtained from the participants’ parents.
Consent for publicationWritten consent for publication of the case details together with imaging have been obtained from participant‘s parent.
 World Journal of Pediatrics2022年12期
World Journal of Pediatrics2022年12期
- World Journal of Pediatrics的其它文章
- Editors
- Information for Readers
- Prevalence of developmental dyslexia in primary school children: a protocol for systematic review and meta-analysis
- Instructions for Authors
- Management of foreign bodies ingestion in children
- Implementation experience of a 12-month intervention to introduce intermittent kangaroo mother care to eight Chinese neonatal intensive care units