
局域表面等离激元诱导的光催化机制
2022-02-24 00:32:52芦一瑞张成云张正龙
陕西师范大学学报(自然科学版) 2022年1期
芦一瑞,严 蕾,张成云,孔 婷,张正龙
(陕西师范大学 物理学与信息技术学院,陕西 西安 710119;西安市光信息调控与增强技术重点实验室,陕西 西安 710119)
光激发贵金属纳米颗粒会引起金属中类自由电子的集体相干振荡,形成局域表面等离激元共振[1-4](图1)。此过程可以显著增强金属纳米颗粒对光的吸收,同时在金属颗粒表面产生强局域电磁场(图1c),从而将更多的能量转移到反应体系内[5-6]。等离激元弛豫过程中,产生高能量的电子-空穴对(图1b)。当等离激元能量与反应物能级间隙匹配,通过高能电子向反应物分子的转移,可以实现反应物基态电子的直接激发[7]。等离激元还可以在纳米、亚飞秒尺度实现对热电子产生和转移过程的精确调控,以达到对化学反应的控制[8]。另外,贵金属纳米颗粒的吸收光谱可以覆盖可见光全波段,能有效地将太阳能转化为化学能,为解决能源问题提供了可行之径[9]。
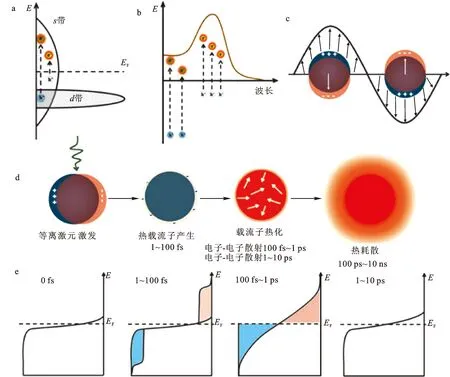
a.金属内电子带内跃迁和带间跃迁[10];b.等离激元金属纳米结构的吸收光谱[11];c.金属电子与光场共谐振荡[11];d.等离激元的非辐射弛豫过程[6];e.等离激元弛豫过程中电子能量分布随时间的演化[6]。
图1d展示了贵金属表面等离激元的激发和非辐射弛豫过程。光场作用于金属纳米颗粒,可在其表面激发局域表面等离激元。等离激元共振能量可以由颗粒的尺寸、形状、材料以及周围的介质加以调控[11]。随等离激元在飞秒尺度下迅速衰减,体系以辐射跃迁的形式释放出光子[12],或以非辐射跃迁的形式将能量转移给载流子,形成热载流子[13]。等离激元模式决定了两种弛豫机制各自占比[14-15]。非辐射弛豫发生在1 fs到100 fs内,可在金属导带内激发电子(带内跃迁)或将其他带上的电子(例如d带)激发到导带(带间跃迁),形成高能电子-空穴对(图1a)。金属颗粒内可能的载流子能量分布依赖于等离激元的能量、颗粒形貌、等离激元模式的对称性、电子结构以及材料态密度分布等因素[16]。通过电子-电子(100 fs~1 ps)及电子-声子(1~10 ps)散射,热载流子的能量转化为晶格热量,产生局域热效应。
热载流子的转移在等离激元催化反应中起到至关重要的作用[11]。热载流子一般分为热电子和热空穴,不同的体系中起主要作用的载流子不同。热电子可以从金属表面转移给表面吸附的分子,也可以直接在反应物分子内产生,分别被称为电子的间接和直接转移过程[10]。由于电子-电子以及电子-声子相互作用,电子的能量会在极短时间内转化为晶格热量,使晶格迅速升温。此过程限制了热载流子的作用,但可以实现金属颗粒高效局域加热,在光化学催化中具有广泛应用[6,17]。
等离激元弛豫过程中产生的热电子能量分布如图1e所示[18]。由于标准等离激元金属的电子逸出功大于等离激元能量ћωP(ωP为等离激元频率),而热电子的能量分布在EF到EF+ћωP之间,无法脱出金属。最初由于等离激元弛豫产生的热电子会通过电子-电子散射过程迅速将能量转移给其他低能量电子,使体系电荷能量呈现费米-狄拉克分布。该过程一般发生在100 fs到1 ps之间。在1~10 ps范围内,由于电子-声子相互作用,热载流子能量损耗,晶格温度上升,引起金属颗粒的局域加热[17,19-20]。等离激元局域电场增强效应及其弛豫产生的热载流子、局域热效应等,使其在光催化领域展现出诸多优势,获得了广泛关注。本文综述近年来在等离激元光催化方面的相关理论和实验研究进展,重点讨论等离激元诱导光催化的微观机理及其应用。
1 等离激元光催化驱动分子反应
1.1 表面等离激元电磁场驱动催化
表面等离激元的激发可直接导致光场能量的重新分布,使金属颗粒表面光子密度增加,进而引起电磁场的显著增强。传统的光催化通过将光子的能量转移给反应物以推动化学反应。与此机制类似,具有更高光子密度的等离激元场可以提高能量转移的速度和效率。如图2b所示,若等离激元场的能量与反应物分子的最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)能级间隙匹配[7],则场与分子间可发生共振能量转移,反应物分子跃迁到激发态势能面上。分子有可能沿激发态势能面继续演化,也可能跃迁回基态势能面,获得与初始状态相比更低的势垒,从而降低反应所需要的能量[16]。
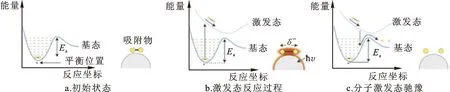
图2 等离激元电磁场诱导分子在金属表面分解反应示意图[16]Fig.2 Schematic of the plasmonic-field-induced dissociation reaction on the surface of a plasmonic metal[16]
在图3所示实验中,分子吸附在铜基底上,构成分子-Cu/Ag体系。在此体系下单个二甲基二硫分子在激光照射下发生分解[21]。该反应中,入射光子能量与分子HOMO-LUMO间隙匹配,实现了分子从基态到激发态的直接跃迁(图3c)。另外,由于分子与金属基底的轨道杂化,分子的HOMO-LUMO能量间隙减小,使电子跃迁需要的能量从紫外光区红移到可见光区,实现了等离激元激发的电磁场驱动的分子反应。

图3 表面等离激元电磁场诱导单个二甲基二硫分子分解[21]Fig.3 Plasmonic-field induced dissociation of a single dimethyl disulfide molecule[21]
1.2 热载流子驱动催化
等离激元弛豫产生的热载流子可以转移到吸附在金属表面的反应物上,从而推动化学反应,将光能转化为化学能。其在光电太阳能、光电探测和光催化等方面都有较为广泛的应用[22]。在两步转移机制中,热电子发生间接转移(图4a)。热电子被激发后,首先和周围的电子相互作用,金属表面电荷能量呈现费米-狄拉克分布,随后由于电子隧穿效应,热电子转移至吸附在金属表面的反应物。诸多研究表明,金属费米面附近的热电子具有很高的转移效率,因此热电子的间接转移被认为是光催化中反应物的主要激发方式。然而,激发光并不能有效控制热电子的能量,极大地限制了对化学反应路径的选择性[23]。
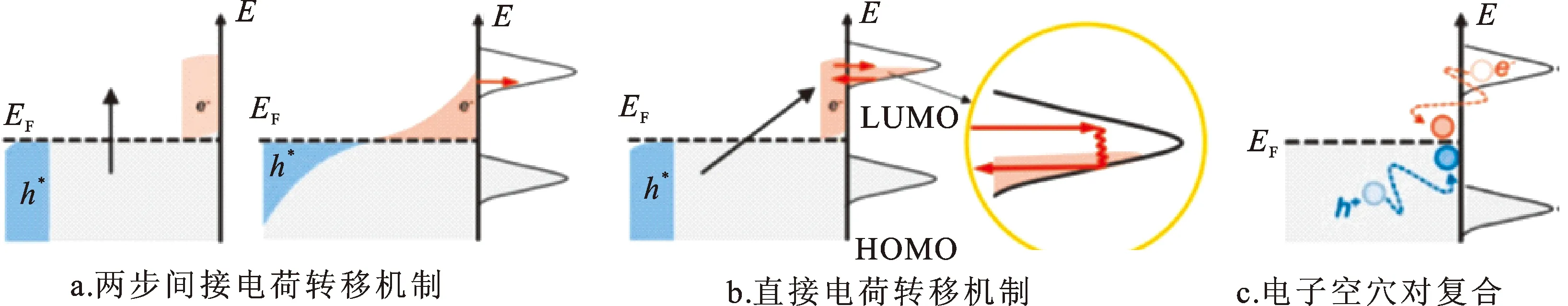
图4 等离激元诱导的热电子转移[11]Fig.4 Plasmon-induced hot electron transfer from the metal to the adsorbed molecule[11]
若吸附物与金属发生较强相互作用,会形成表面杂化态,从而提供等离激元与吸附物直接作用的路径。等离激元通过与表面杂化态的耦合可以直接在反应物空态上产生热电子,在金属表面产生空穴(图4b)。与热电子间接转移相比,由于没有金属表面的电子散射,热电子能量损失减少。然而由于分子-金属结构转移偶极矩较小,且需要形成杂化的表面态,热电子的直接转移的效率较低。另外,热电子的直接转移具有更加严格的激发条件,等离激元共振能量与表面杂化态的能量必须匹配,才能激发电子从金属占据态到反应物空态的跃迁[12]。
1.2.1 热电子间接转移
许多研究认为,热电子的间接转移机制在等离激元光催化中占主导地位[24-25]。如图5a所示,通过高真空尖端增强拉曼光谱(HV-TERS)对表面等离激元诱导对硝基噻吩(PNTP)原位化学反应,发现等离激元诱导PNTP分子的催化反应机理主要是由于热电子的间接转移,而对温度环境不敏感。在金属针尖和衬底间的热点处等离激元共振产生的高密度热电子,会通过间接转移的方式转移到吸附在金属表面PNTP分子的未占据轨道上,诱导其发生聚合反应[26]。

a为HV-TERS揭示的热电子驱动PNTP的化学反应[26];热电子驱动的Au纳米颗粒表面H2解离[27]:热电子激发(b),热电子从金颗粒转移到分子的LUMO轨道(c),H2分子分解反应的机制(d);热电子诱导的O2分解反应[23]:吸附于Ag表面的O2的态密度图(e);O2和的势能面(f);热电子诱导O2分解反应的机理(g)。
图6展示了利用数值模拟银链等离激元诱导的氢分子分解,阐明了热电子向氢分子间接转移导致氢分子分解的过程[28]。在飞秒激光的激发下,银链上的等离激元诱导吸附的氢分子发生分解(图6a)。如图6b—e所示,吸附在银链上的氢分子仅在特定的激光频率下分解,说明分子分解是由光激发银链的等离激元所导致。通过对氢分子周围电荷的计算,可以观察到电子在氢分子周围的聚集,并在20 fs左右达到一个短暂的带负电荷的氢分子状态,随后氢分子开始分解。图6f展示了基态轨道的占据数随时间的变化以及对应态的波函数。从Kohn-Sham(KS)态含时演化图可以看出,氢分子反键态的占据数随演化持续增加,进一步证明了热电子由银链转移到了氢分子的反键态。以上现象表明了典型的电子间接转移机制:银链上的电子被激光激发,然后转移到氢分子的反键态,引起了氢分解[23,27-29]。

a为等离激元诱导氢分解示意图;包含6个银原子银链上氢分子分解:模拟选用的高斯形激光脉冲(b),不同频率激光激发下氢分子键长变化(c),不同场强下氢分子键长时间演化(d),氢分子周围电荷分布(e),基态下KS态随时间的演化(左)以及对应态的波函数(右)(f)。
1.2.2 热电子直接转移
金纳米颗粒等离激元诱导水分解的理论模型及其电子-原子核超快动力学的研究可以证明热电子的直接转移机制(图7a)。吸附在金纳米球上的水分子可在场强Emax=16 V/nm、能量ћω=2.62 eV的脉冲激光激发下发生分解(图7b),此激发频率取自金球等离激元的共振吸收峰。在水分子分解的过程中,氧原子基本保持不动,其中指向金球的一个氢原子仅发生振动,另一个氢原子先振动然后分解。即水分子中一个H—O键保持,一个H—O键断裂,在30 fs内分解为一个氢氧基和一个氢离子(图7c)。相比之下,若将水分子直接置于此电场下则不会发生分解,可以证明金球上的等离激元导致水分解[30]。
图7d展示了不同尺寸金球吸收谱与水分子分解速率的波长依赖性,进一步证明了等离激元模式与水分子分解的关系。对于直径为1.6 nm的金球,在光吸收强的激光频率下水分解速率比较高,吸收峰和分解速率峰值基本对应。然而对于直径为1.9 nm和2.1 nm的金球,由于尺寸效应,光吸收峰在2.30 eV处变弱,在其他频率下产生较强的吸收,水分解速率与其呈现不同趋势。从灰色区域对比可看出,直径为1.9 nm的金球在吸收峰较低的频率却有较高的水分解速率。直径为2.1 nm的金球在2.62 eV处的吸收峰值达到最大,水分子分解速率却与2.30 eV基本持平。说明水的分解不仅与光吸收有关,而且与特定模式的等离激元相关联。
对等离激元模式的进一步分析发现,共振能量为ћω=2.36 eV的等离激元诱导的电荷密度振荡周期为奇数,称为奇模式;能量为ћω=2.62 eV的等离激元为偶模式。当水分子吸附在金球上时,奇模式比偶模式在水分子周围产生更多的诱导电荷。图7e所示KS态的占据数表明,当偶模式等离激元(ћω=2.62 eV)被激发时,费米能级附近轨道占据数有很强的振荡,称为电子振荡。电子从深能级范围-1.43~-0.72 eV 的轨道跃迁到1.40~1.50 eV,称为反转行为[31]。费米能级附近电子振动行为来源于纳米颗粒表面附近电荷密度振动,而反转行为是由于等离激元弛豫产生的热电子与热空穴。与偶模式相比,在奇模式2.36 eV的激发中,KS轨道占据数在费米能级附近的振荡很弱,主要是由奇模式吸收强度小导致。电荷反转主要是从-1.90~-1.50 eV 的能量范围到0.58~0.79 eV。对于偶模式,电荷主要反转到1.40~1.50 eV的能量区间。可以看出,等离激元诱导热电子的能量和吸附物空态能量的匹配对电子的直接转移起到至关重要的作用。

a.金颗粒表面等离激元诱导水分解示意图;b.外加电场演化图;c.分子的分解过程;d.水分子在不同频率下分解速率与光吸收谱的对比,分别对应三组金球直径;e.连续平面波激发下KS态的占据数随时间的演化,以及相应能量下水分子的局域态密度,黑色实线代表激光场振荡。
研究者通过实验观测到等离激元诱导金属-反应物接触面上的电子转移跃迁[32],等离激元弛豫直接激发金属表面的电子到反应物上,证实了热电子的直接转移过程(图8)。图8a为CdSe-Au纳米线的透射电子显微镜(TEM)表征图,可以看出金纳米颗粒附着在CdSe两端,呈哑铃状。CdSe-Au纳米线在三氯甲烷溶剂中的吸收谱如图8b所示,CdSe-Au纳米线在480 nm和582 nm附近分立的吸收峰分别由1πe和1Σ激子能带贡献。图8c展示了光激发CdSe内电子带间跃迁以及等离激元弛豫实现金纳米颗粒电子被直接激发到CdSe的1σe能带上。从图8d可以看出,CdSe两端加上金纳米颗粒后,吸收峰范围明显增宽,且接近红外区,起始吸收波长约在1 450 nm (0.85 eV)处。此现象可能是由于CdSe和Au间存在强电子轨道耦合,从而引起了新的等离激元弛豫路径。这种机制使得热电子被直接激发到CdSe中,Au中仅产生空穴,实现了电子的直接转移。
2 等离激元驱动纳米结构的光催化
除了驱动分子反应,贵金属纳米颗粒上的等离激元在驱动纳米结构光催化反应中也有广泛应用。等离激元的高空间局域性和超短时间尺度为精确控制纳米材料的生长和转变提供了有效途径。光激发的局域表面等离激元诱导吸附在金属纳米颗粒表面的分子发生催化反应,形成的金属原子或其他产物发生沉积,导致纳米材料生长。对于纳米材料的转变,等离激元的直接催化目标是无机纳米颗粒,这开辟了等离激元驱动反应的新领域。

a.CdSe-Au纳米线TEM典型表征图;b.三氯甲烷溶剂中CdSe纳米线,CdSe-Au纳米线和CdSe量子点-Au二聚体吸收谱,灰色虚线是CdSe-Au纳米线和CdSe量子点-Au二聚体吸收谱差;c.CdSe-Au纳米线电子结构及电子直接转移的示意图;d.首个激子峰位分别在555 nm、582 nm、605 nm的CdSe-Au和CdSe吸收谱。
2.1 等离激元驱动的纳米材料生长
等离激元诱导的局域强电磁场通过有效加速氧化还原反应使金属优先在热点处沉积,从而实现对金属生长在空间上的控制。图9a展示了表面等离激元诱导银纳米三角片的生长[33]。表面等离激元驱动金纳米片的生长体现了等离激元对贵金属纳米颗粒晶体结构的催化作用(图9b—f)。由于结构的内在差异,可以通过调节热电子的输运来调节纳米孪晶的生长动力学[34]。另有研究发现,表面活性剂聚乙烯醇吡咯烷酮(PVP)不仅有镜面阻断的作用,而且还能促进金属表面热电子的累积。在热电子的作用下,还可实现Pt在金纳米棒尖端的靶向沉积(图9g—h)。纳米棒表面热电子催化了Pt0的还原,促进金纳米棒尖端Pt0外延生长[35]。此外,等离激元诱导的电磁场空间分布不均匀导致Pt在金纳米棒特定位置的靶向沉积,实现了Pt的纵向生长。利用表面等离激元加热效应,Cao等设计了一种在室温下快速加热和冷却材料的思路,实现了硅纳米线在室温下的可控生长[36](图9i)。

a.等离激元诱导银纳米三角片在溶液中生长的示意图和样品的TEM图[33];b—f为表面等离激元诱导金纳米片的生长[34];生长后纳米结构的SEM图(b)和Au-(c)和12C14N(d)元素分布图;g-h为表面等离激元诱导金纳米棒表面Pt4+还原为Pt0,Pt修饰金纳米棒的结构示意图和元素分布图[35];i为表面等离激元热电子诱导硅纳米线生长[36]。
2.2 等离激元光催化驱动的纳米晶体转变
等离激元纳米颗粒除了与分子相互作用催化相关化学反应外,其在纳米材料相关反应中起到的催化作用也受到了广泛的关注。等离激元的空间局域特性为精确调控纳米材料的生长和转变提供了可行之径。图10a为等离激元驱动稀土掺杂发光纳米晶体转变的示意图,利用金属纳米结构可实现多晶氟化物向单晶氧化物的转变[37]。如图10b所示,金纳米颗粒等离激元共振弛豫产生的热电子和热效应协同作用促进了晶体材料的转变。从图10c颗粒经激光辐照前后原位荧光光谱的变化可观察到,等离激元驱动晶体转变依赖于激光辐照的波长。532 nm的辐照光可在25 ms内快速获得单晶氧化物,而633 nm的辐照光却无法实现晶体的转变,这是由于金颗粒的等离激元共振峰位于532 nm附近,在此波长处具有最大的消光截面,能有效地激发金属表面电子的集体振荡。此外,在低温环境中也可以实现表面等离激元驱动的纳米晶体转变,突破了传统方法对高温环境依赖这一技术的瓶颈(图10d)。

a.表面等离激元诱导多晶NaYF4向单晶Y2O3快速转变的示意图;b.等离激元驱动纳米结构转变的动力学过程以及作用前后多晶氟化物和单晶氧化物的STEM图对比;c.NaYF4:Eu3+@Au在不同波长激光辐照前后的荧光光谱图;d.晶体转变所需时间随温度的变化。
等离激元高效的局域热效应被认为是催化晶体转变的主要机制,因此有效提高体系的热利用率,增强等离激元的光热效应是提高晶体转变效率的关键。另有研究发现,在自组装的金纳米岛表面引入Al2O3薄层可以有效地增强光吸收和热利用(图11a),相比于纯金纳米岛体系,热捕获结构可使晶体转变速率提高近十倍[38],从而增强等离激元驱动晶体转变的光热效应,并且在低温环境中也能实现相对较高且稳定的光热转换效率。图11a为等离激元驱动晶体转变的热捕获体系示意图,Au纳米岛为等离激元光热基底,在其表面引入热捕获层Al2O3,NaYF4多晶在激光照射下可快速转变为Y2O3单晶。图11b显示了重叠区域热捕获结构的截面示意图。对热捕获结构进行FIB切割,切割后产物薄片的元素分布图(图11c)表明厚度为30 nm的热捕获层Al2O3均匀地沉积在Au纳米岛(AuNIs)的表面上。对于热捕获体系,由于有效折射率的增加,光吸收截面随着Al2O3层厚度的增加而逐渐增加,并且Au纳米岛产生的大量热量主要转移到介质层Al2O3上;另外,位于光斑附近更多的AuNPs产生的大量热量通过Al2O3层转移到晶体上,导致晶体温度迅速升高,实现了晶体转变效率的提升。总之,由于增强的光吸收和热利用,热捕获结构可增强等离激元驱动晶体转变的光热效应。
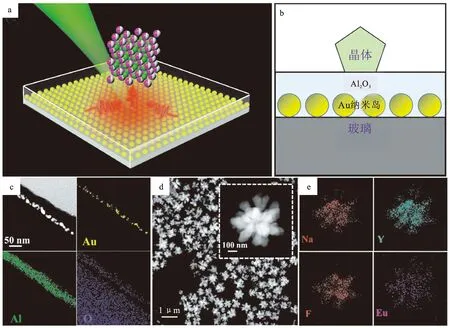
a.热捕获体系示意图;b.热捕获体系截面示意图;c.热捕获结构截面的HAADF-STEM图和EXD元素分布图;d.NaYF4:Eu3+亚微米晶体的HAADF-STEM图;e.单个亚微米晶体的元素分布图。
3 结语
相比传统的光催化,贵金属纳米颗粒的等离激元催化具有很多优势:金属颗粒的吸收波长可以被调控到可见光范围,有利于充分利用太阳能,对环境更加友好;等离激元的模式可以由外加光场调控,进而实现对反应的选择性催化;等离激元弛豫产生的热载流子可以高效地诱导反应发生;热载流子弛豫可以实现对颗粒的局域加热,为反应提供能量。
与此同时,等离激元光催化的研究也面临诸多挑战:在理论研究方面,不同机制发生的时间和空间尺度可能存在较大差异,导致难以建立精确描述等离激元催化过程的一般模型;分子动力学研究尺寸和能量转移机制研究尺寸也不同,使分子催化中能量转移过程难以被计算模拟。因此,需要建立一种具有多尺度鲁棒性的模型来进行更深层的理论研究。另外,反应体系激发态精准模型的建立仍需进一步的理论突破。在具体的催化机制探究上,由于反应与催化剂性质(组成结构、尺寸形状、表面化学性质等)、反应物性质(相、组成等)、激发源性质(波长、强度、流量)等多方面因素相关,因此如果判断某种非热激发机制为催化反应主导机制,该机制需要对所有不同情况做出合理解释。并且对于一种机制的描述既需要指明作用物(如热载流子、强电场),也需要讨论具体作用过程。此外,等离激元热效应引起的局域温度难以被精确测量或计算,然而即使很小的温度误差都会导致非热催化机制作用被极大的低估或高估。在具体应用上,等离激元光催化距离实现工业化大规模应用还有很多亟待解决的问题[39]。
猜你喜欢
农村青少年科学探究(2020年5期)2020-08-18 02:20:52
陶瓷学报(2019年5期)2019-01-12 09:17:34
电子测试(2018年18期)2018-11-14 02:30:34
新民周刊(2018年8期)2018-03-02 15:45:54
饮食科学(2017年12期)2018-01-02 09:23:20
三峡大学学报(自然科学版)(2017年1期)2017-03-20 15:30:23
中国资源综合利用(2016年9期)2016-01-22 08:35:22
中国医学装备(2015年10期)2015-12-29 12:00:24
少儿科学周刊·少年版(2015年1期)2015-07-07 21:57:30
电测与仪表(2015年7期)2015-04-09 11:39:50
