
Molecular and genetic markers in hepatocellular carcinoma: In silico analysis to clinical validation (current limitations and future promises)
2022-02-24 05:59SarahElNakeep
Sarah El-Nakeep
Sarah El-Nakeep, Gastroenterology and Hepatology Unit, Department of Internal Medicine,Faculty of Medicine, Ain Shams University, Cairo 11591, Egypt
Abstract Hepatocellular carcinoma (HCC) is the second cause of cancer-related mortality.The diagnosis of HCC depends mainly on -fetoprotein, which is limited in its diagnostic and screening capabilities. There is an urgent need for a biomarker that detects early HCC to give the patients a chance for curative treatment. New targets of therapy could enhance survival and create future alternative curative methods. In silico analysis provides both; discovery of biomarkers, and understanding of the molecular pathways, to pave the way for treatment development.This review discusses the role of in silico analysis in the discovery of biomarkers,molecular pathways, and the role the author has contributed to this area of research. It also discusses future aspirations and current limitations. A literature review was conducted on the topic using various databases (PubMed, Science Direct, and Wiley Online Library), searching in various reviews, and editorials on the topic, with overviewing the author’s own published and unpublished work.This review discussed the steps of the validation process from in silico analysis to in vivo validation, to incorporation into clinical practice guidelines. In addition,reviewing the recent lines of research of bioinformatic studies related to HCC. In conclusion, the genetic, molecular and epigenetic markers discoveries are hot areas for HCC research. Bioinformatics will enhance our ability to accomplish this understanding in the near future. We face certain limitations that we need to overcome.
Key Words: Hepatocellular carcinoma; In silico analysis; Bioinformatics; Biomarkers;Molecular pathways; Genetics; Epigenetics
lNTRODUCTlON
Hepatocellular carcinoma (HCC) is the most common primary liver cancer. HCC is the sixth most prevalent cancer, and the second leading cause of cancer mortality[1]. The incidence of HCC in different countries is related mostly to the geographic prevalence of certain risk factors such as chronic hepatitis B and C, aflatoxins, and alcoholism[2,3]. HCC causes annual morality exceeding 700 000 cases[4], and high recurrence rate after treatment, with overall 5-year survival (< 50% of cases)[5], and even lower numbers reach the endpoint of 10-year survival (< 10% of cases), despite aggressive treatment[6].
Cirrhosis can proceed to HCC in 5%–30% of patients after an average duration of 5 years[7]. Most HCC cases arguably occur on top of cirrhosis, but we cannot ignore the 20% of cases that occur without any preceding cirrhosis. Thus, cirrhotic and noncirrhotic causes of HCC are explained by different pathogenic mechanisms[8,9].
A debate has arisen about the relation of increased incidence of HCC among patients receiving new direct-acting antivirals (DAAs) for hepatitis C treatment, but recent studies have shown that the risk ofde novoand recurrent HCC with DAAs is actually lower than without, although not completely abolished[10,11].
Many HCC studies use bioinformatics as a method to determine the molecular pathways affected by HCC, along with the genetic and epigenetic control of those pathways. These proteomic and genomic studies are the future of personalized medicine, where precision therapy could offer patients a management plan specified for their individual mutations, with high curative capabilities. We have visualized how intercepting a molecular pathway as in sorafenib, a multikinase inhibitor that enhances apoptosis, could increase the survival of advanced HCC by several months,but unfortunately, recent studies have shown that resistance to the drug is evolving[12,13].
The Cancer Genome Atlas Research Network has conducted a study project on 33 cancers with poor prognosis, provided that there were suitable available tissue samples for experimental validation through antibodies’ multiplex analysis, and they included HCC among other cancers. The HCC study included 363 cases for wholeexome sequencing and 196 for further proteomic, epigenetic and DNA-methylation analyses. They found that the molecular pathways most affected in HCC are those that deal with the following: cell proliferation, differentiation, growth, apoptosis, and immortalization (through telomerase)[14].
In this review, I explore the steps for validation of molecular markers, with the limitations encountered to validate a novel biomarker, the research in molecular pathways related biomarkers, the role of bioinformatics in the discovery of those pathways, and future aspirations.
lMPORTANT QUALlTlES lN DlAGNOSTlC AND PROGNOSTlC MARKERS THAT HAVE TO BE MET
The early diagnosis of HCC is a crucial issue, as all of the available curative measures are only effective in early stages of cancer (liver resection, liver transplantation,radiofrequency ablation). They are curative in early the Barcelona Clinic Liver Cancer stages (0 and A), where the size of the tumor does not exceed 5 cm in its largest diameter in one nodule, or the size does not exceed 3 cm in three nodules (Stage A)[15]. Thus, early screening of HCC is an effective tool for both early detection and treatment, which increases overall survival and yields better prognosis. Unfortunately,the screening process of HCC suffers a huge limitation, which is the low sensitivity of its most accepted biomarker, -fetoprotein (AFP).
So, the current European Association for the Study of the Liver (EASL), and the American Association of Study of Liver Disease (AASLD) guidelines recommend the following; due to cost-effectiveness, abdominal ultrasound should only be used in the screening process, while AFP is limited to the diagnosis or screening of at high-risk populations. Where we find AFP sensitivity reaching as low as 20% positivity in early stages of cancer, with fluctuating levels of the biomarker in cirrhotic patients due to other reasons, causing further confusion in reaching the diagnosis[16-18], AFP is removed from the screening assessment guidelines altogether, as mentioned above.
It is important to note that, in Japan, the at-risk populations for HCC are still screened by 3-mo abdominal ultrasound, and AFP, in addition to another two biomarkers,Lens culinaris-agglutinin-reactive fraction of AFP, and PIVKA-II (proteininduced by vitamin K absence or antagonist-II). All these are included in the Japanese insurance plan of at-risk populations[19]. Other markers considered for HCC diagnosis are: Dickkopf-1, which is a good biomarker for HCC with negative AFP[20],and des--carboxy prothrombin, which is directly correlated with tumor size and has higher sensitivity than AFP, so can be used in screening more effectively[21]. Unfortunately, none of the aforementioned biomarkers reaches the final acceptance to be added to any of the clinical practice guidelines for HCC due to cost-effectiveness,difficult availability, or high variability across studies.
I shared in the research work of determining some of the cost-effective biomarkers that are both cheap and effective, for establishing the diagnosis and staging of HCC,including a study on Golgi protein (GP)73, where the combined sensitivity of both AFP and GP73 was 84.4% and specificity of 95.6%[22]. Our results were similar to the meta-analysis of the diagnostic accuracy of GP73 in HCC, where combined GP73 and AFP had pooled sensitivity of 87% and pooled specificity of 85%[23].
AFP is the only widely validated biomarker for HCC diagnosis, and prognosis in most clinical guidelines despite its limitations. To overcome its limitations we are still searching for a new biomarker. This is an ongoing process, requiring computational,experimental, and clinical validation. Figure 1 shows the most important factors that are required in an effective diagnostic and prognostic biomarker.
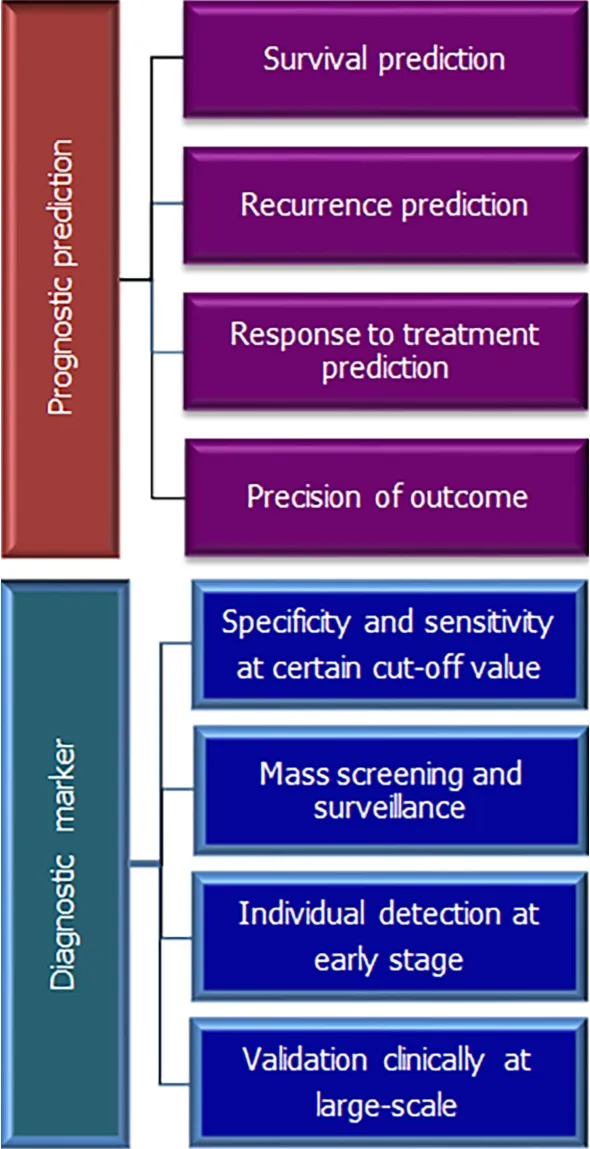
Figure 1 Showing important features of prognostic and diagnostic markers.
STEPS FOR VALlDATlON FOR A NEW BlOMARKER
The only approved biomarker for diagnosis by both the American and European guidelines is AFP. Other biomarkers are approved in Japanese and Korean guidelines as mentioned earlier. AFP, the biomarker that stood the test of time, has its own problems as low sensitivity making it weak as a surveillance method, which caused the ASLD and EASL to remove it from the screening of HCC except for high-risk populations[24,25]. Searching for a new biomarker is an ongoing process, which needs computational, experimental, and clinical validation, as shown in Figure 2.

Figure 2 Pathway for validation of the biomarkers in hepatocellular carcinoma.
The novel biomarkers discovered through bioinformatics analysis usually pass through different steps of validation. First, computational validation(in silicovalidation), through assessing correlated genes, then by statistical analysis of different genetic expressions[26]. Hence, the most statistically significant biomarkers, with a plausible molecular pathogenic background, will pass to the next stage. Experimental validation on the HCC tissues on resected tumor from patients or experimental laboratory cells as HeLa cells (in vitrostudies). Later, clinical validation in the sera of patients with established diagnosis to determine the actualin vivopredictive diagnostic and prognostic capabilities of the biomarker. In this stage, we calculate the diagnostic test accuracy of the biomarker through identifying its specificity, sensitivity,and area under the curve (AUC), along with other important related parameters.
Diagnostic biomarkers should have a correlation with a clinical endpoint to be used in clinical trials, whether to help assessing this clinical endpoint or relate to it, as a surrogate marker.Then, external validation is examined through wide-scale studies,for the most acceptable biomarker in sensitivity and specificity. These studies must be done on variable and random populations (different ethnicity, gender, age groups,stages of the disease,etc.). When the biomarker reaches this stage of diagnostic accuracy validation, and proves to be cost-effective, then it can be added to clinical practice according to the level of evidence provided (the type of studies conductedi.e.,cohort, randomized controlled trial, case–control,etc., and the size of the population examined)[27-29].
The steps for validation of a miRNA biomarker are[30]: (1) Data processing and screening of differentially expressed miRNAs; (2) Construction of the miRNA signature; (3) Confirmation of the miRNA signature; (4) miRNA signature validation using the OncomiR database and Gene Expression Omnibus (GEO) dataset; (5)Functional analysis; (6)In vitroanalysis; (7) Testing on patients sera for diagnostic test accuracy; (8) Wide-scale clinical validation; and (9) Adding as a biomarker to the HCC diagnosis guidelines according to the level of evidence.
DlSCOVERlNG NEW BlOMARKERS: MOLECULAR PATHWAYS DlSCOVERY AND DETERMlNlNG OF THE GENETlC-EPlGENETlC-PHENOTYPlC LlNKAGE THROUGH CLlNlCAL STUDlES
HCC is a cancer of poor prognosis, especially when discovered in late stage, which is usually the case, due to lack of early detection by biomarkers, lack of effective chemotherapeutic treatment, and limited molecular target treatment. Understanding the molecular and genetic pathways is vital to overcome these obstacles, and reach better prognostic outcomes. Recently, an accumulation of data regarding genetic and epigenetic biomarkers became available, for bothin vitrolaboratory analysis andin silicoanalysis[31].
Many pathways have been critically assessed as the key element of early diagnosis of HCC or as the key predictive outcome (whether metastasis, relapse or complete recovery) after curative interventions. These pathways include cellular effects, such as:cell proliferation, growth, differentiation and immortality.Moreover, disturbance or mutation affects functions such as: vascular angiogenesis, inflammatory response,programmed cell death and autophagy. Formulation of drugs that could intercept these molecular pathways to establish a treatment plan with good prognostic capabilities is under investigation[32].
Autophagy pathway in HCC: published and unpublished work of the author
Autophagy is defined as the degradation of cellular components by lysosomal fusing with autophagosomes, and forming autolysosomes, as a homeostatic regulation for aging, stress, immunological response, or anticancer response. The role of autophagy in HCC is a complicated one. Whereas basal autophagy is responsible for anticancer protection of the organ, as carcinoma progress to a late stage, autophagy helps the cancerous tissues’ survival and growth. Autophagy genes and their regulatory proteins linked to HCC include,Beclin-1,ATG5andATG7. They control many molecular pathways such as: phosphatidylinositol-4,5-bisphosphate 3-kinase PI3K/AKT/mTOR, ERK/mitogenactivated protein kinase (MAPK), and apoptosis p53 pathways among others[33-36].
Our research on this pathway, linking the autophagy control ofATG-4BmRNA expression through noncoding miRNA-661 through bioinformatics methods proved to be of a clinical value after clinical validation. We found that combination of both biomarkers had specificity of 82.1% and sensitivity of 100%, especially in early HCC.The prognosis in the form of tumor-free survival was improved with the decline in the serum level of the two biomarkers as proved by multivariate analysis[37].
Hepatitis B and C associated HCC and the molecular pathways discovered:published work
Hepatitis C virus (HCV) and hepatitis B virus (HBV) are the most common risk factors associated with HCC[38]. HBV is responsible for about half of HCC cases worldwide,in addition to most of the childhood associated HCC[7].
In a recent study, the researchers performed bioinformatics analyses using data from GEO database, to show the possible molecular pathways, which cause HBV to induce HCC. They formed heatmaps of the top 50 downregulated genes, and the top 50 upregulated genes associated with HCC occurrence. They found that there are six genes most significantly controlling the following pathways: carbon, certain amino acids, and retinol metabolism. They presented the molecular and cellular cycle pathways through the protein–protein interaction networks[39]. Furthermore, HBVrelated HCC is linked to mutation of theTP53gene, along with viral genetic integration with the host DNA[14].
As for HCC associated with HCV, while using hierarchical clustering of the hub genes[40], the authors found overexpression of three genes: cyclin B1 coding gene,kinesin family member 20A coding gene, and hyaluronan mediated motility receptor coding gene. These were associated with decreased survival in patients with HCVassociated HCC[41].
Similarly, our team conducted a study about the relation ofIL-28genetic polymorphism and HCC associated with HCV, in the era of interferon treatment of HCV infection. We found that the T allele was higher in both chronic liver disease (CLD)and HCC groups, with prevalence of 50% and 70%, respectively. As compared to the C allele, where the prevalence in CLDversusHCC groups was 30% and 50%,respectively, but the differences between the groups were not significant[42]. Our results were similar to a recent study conducted on the Chinese population, which found that the T allele was associated with a higher risk of HCV-related HCC[43]. A recent meta-analysis found a strong correlation betweenIL-28Bgenetic polymorphism and HCC association with HBV or HCV infection, where CC and TT genotypes of certain single nucleotide polymorphisms (SNPs) ofIL-28Bwere protective against HCC occurrence[44].
In addition, a bioinformatics study found that theIL-28Bgene has a relation to HCC recurrence through gene expression profiles on 20 HCCversus91 CLD samples,further researchedin silicoby gene set enrichment analysis and one-way hierarchical clustering for microarray analysis. They found on subsequent clinical validation in 183 HCC patients that certainIL-28Blocus SNPs are associated with HCC recurrence [45].
Role of noncoding RNAs as epigenetic biomarkers for HCC: including published work
Both long noncoding RNAs and miRNAs are considered noncoding chromosomal regions, originating in the introns of the chromosomal DNA. They are responsible mainly for the control of the exons’ stimulation–inhibition process[46]. Exons are the chromosomal blocks of DNA responsible for encoding proteins. The noncoding RNAs have many functions including: controlling protein metabolism, and maintaining chromosomal structure, besides segregation through telomerase generation[46,47].
As an example, the role of miRNA-20a in controlling the mRNA ofCyclinD1(a proto-oncogene) was studied using bioinformatics prediction methods through matching the seed region of the miRNA with the chosen sequence, to predict related miRNA targets. This oncogene is responsible for controlling progression of G1 to S phase, and hepatocellular growth, through regulation of the Wnt signaling pathway.Later, this was confirmed by experimental validation in HepG2 cells[48]. Table 1 shows the molecular pathways.
Also, miR-1180-3p is upregulated in HCC and associated with increased tumor proliferation, resulting in poor survival. Functional computational analysis and KEGG pathways maps showed that this epigenetic marker is linked to MAPK pathway regulation, in addition to control of cellular proliferation, apoptosis and differentiation[49]. Table 1 shows the molecular pathways.
Our team has published work on the relation between different miRNAs and oncogenes and HCC, especially those associated with HCV, and comparing their diagnostic efficacy to that of the established marker AFP. For example, autophagy genetic markers are correlated with miRNA-661, as mentioned earlier. lncRNA-UCA1(urothelial carcinoma associated 1), c-JUN (cellular jun proto-oncogene), miR-143 and miR-550a were studied in the serum of HCV-infected HCC patients[37,50,51]. lncRNAUCA1 had a sensitivity of 91.4% and specificity of 88.6%, while c-JUN had a sensitivity of 91.4% and specificity of 91.4% with AUC of 91%[51]. Also, miR-550a had an inverse relation with miR-550a with sensitivity of 91.89% and specificity of 90.24%, while miR-143 did not show any relation to HCC occurrence[50].
Role of telomeres in HCC initiation and prognosis: work in progress
Telomeres function mainly in capping the chromosomal end to protect it from damage. They consist of nucleoprotein repeats. Telomeres can be transcribed into long noncoding RNAs, thus having an epigenetic control on the telomere homeostasis and telomerase enzymatic activity. Telomeres that bear such functions are called telomeric repeat-containing RNA (TERRA)[52,53]. The role of TERRA in HCC prognosis has been recently studied; it is downregulated in HCC and causes poor prognosis due to metastasis and cell growth, as studiedin vivoandin vitro[54]. Through bioinformatics analyses, several regulatory protein motifs (regulating TERRA) at the end of chromosomes were identified and confirmed through experimental siRNA transcription on HeLa cells, when transfected[55]. Determination of the mechanisms of control of telomere homeostasis and telomerase enzyme will enable researchers to discover drugs that could modify this pathway, in order to cure cancer proliferation,and metastasis (Figure 3). I am involved in ongoing research in this area.
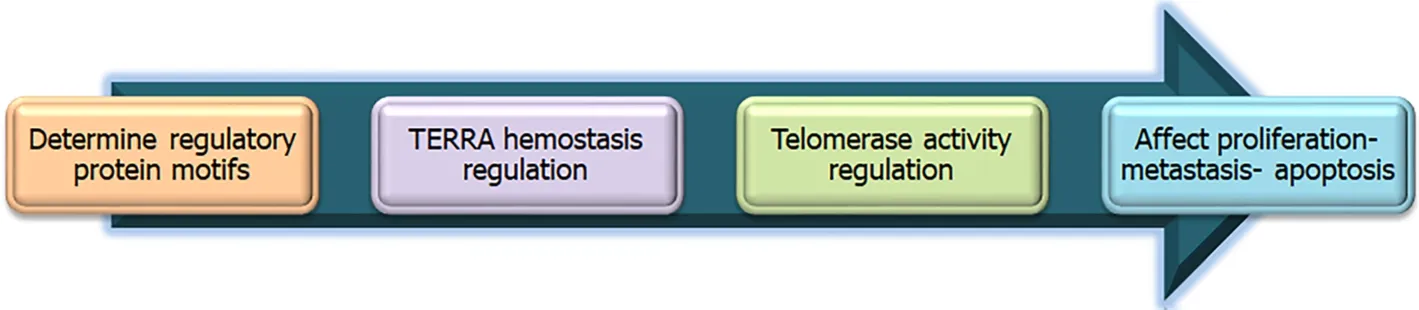
Figure 3 Proposed mechanism of drug development using bioinformatics and molecular knowledge about telomere homeostasis.
Other important molecular pathways
Other important molecular pathways that are studied in HCC are shown in Table 1. (1)Proliferation pathway is enhanced through the inhibition of various transcription factors (TFs). TFs present a form of differentiation therapy, which decreases cancer growth[56]. (2) Cellular growth in HCC: growth factor dysregulation causes disturbance in hepatocyte growth, and is considered as a treatment option for HCC[57]. (3)Angiogenesis in HCC: diagnosis and prognosis of HCC have different associations with various growth factors, including vascular endothelial growth factor (VEGF),epidermal growth factor, transforming growth factor,etc. (4) Inflammatory response:for example, the effect of the interleukin pathway, and chronic inflammatory response in chronic hepatitis or steatohepatitis could result in activation of this pathway. And(5) Cell cycle progression: as mentioned earlier in control of cyclin D1, this could also form a suitable drug target.
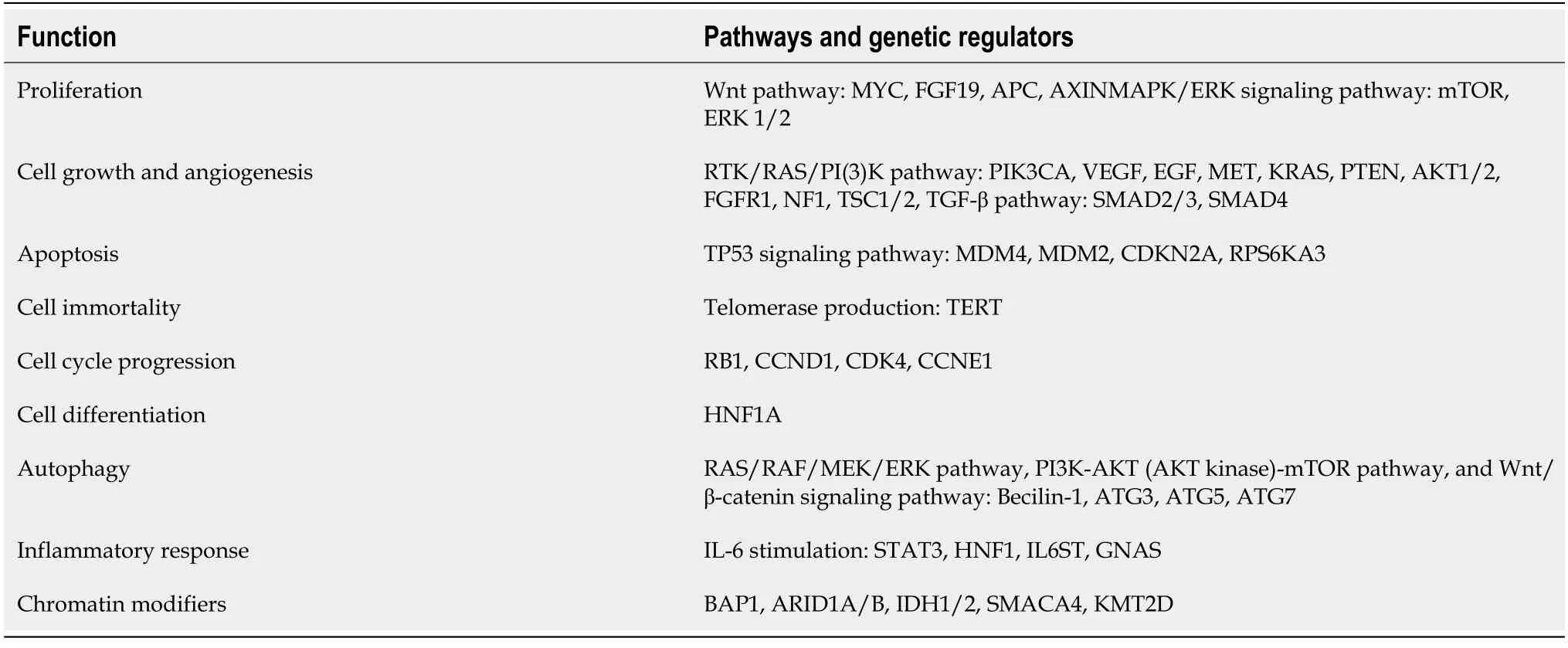
Table 1 Molecular pathways affected in hepatocellular carcinoma and their related protein-coding genes[14,33,34] and KEGG pathways database[35]
COMPUTATlONAL METHODS USED lN MOLECULAR PATHWAY DlSCOVERY
Interactive networks formed by data mining
Genetic networks formed through data mining are formed of two types: supervised learning, which mainly investigates data through statistical analysis of the patterns of coexpression presented in different genes; and unsupervised learning, which mainly deals with the discovery of genetic signatures to predict occurrence of certain diseases[58]. Both are considered methods of artificial intelligence and machine learning.Identifying the coexpression of genetic patterns for diagnosis and prognosis, through machine learning, could help formulate personalized therapeutic targets and advance precision medicine[59]. Figure 4 shows the pathways of bioinformatics analysis, and the general directions aimed in usingin silicoanalysis.
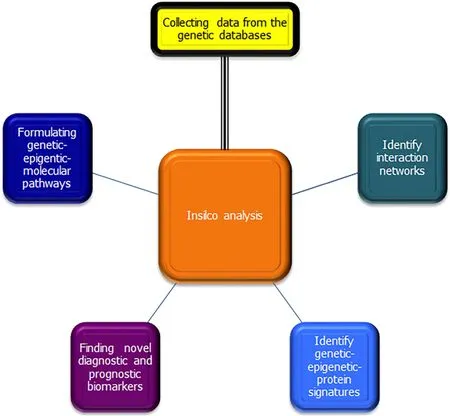
Figure 4 Pathways and aims of bioinformatics analysis.
Forming a miRNA signature
miRNA signature is a group of miRNAs that act collectively as one diagnostic or prognostic biomarker for a certain disease. By using the most relevant and lowest number of miRNAs to achieve the highest possible sensitivity and specificity of this biomarker in diagnosis or prognosis, we can create a relevant signature. Recently a group of scientists used support-vector-machine-based technology to assess the relation of miRNA signatures with clinical staging of HCC. The results showed 23 miRNAs with collective high sensitivity and specificity in differentiating early from late HCC, while seven miRNAs helped to determine the prognosis and survival in HCC patients[60].
Forming of prognostic biomarker coexpression signatures in HCC
Genetic or protein signatures formed by alignment of different sequences, preferably through multiple sequence alignment, could provide information about the "most conserved" sequence in a protein or gene or miRNA, through comparison between genes inherited by different species with a common ancestor (homologs), including similar genes in different species (orthologs), or different genes in the same species(paralogs). Coexpression signatures might help to categorize proteins or genes in different familial sets to predict their prognostic effect. There are different types of coexpression signatures including patterns, fingerprints and profiles[61-63].
A group of researchers collected different known HCC prognostic genes from various genetic and oncological databases, then through Lasso-Cox modeling a single prognostic signature, composed of the five genesCCNB2,DYNC1LI1,KIF11,SPC25andKIF18A, was tested in HCC tissues from patients by immunohistochemistry against HCC survival[64]. Another group of researchers found a single 14-gene signature for the prediction of HCC outcome[65].
A recent proteomic study used data mining to examine a new prognostic predictor protein signature. They found that four proteins, proliferating cell nuclear antigen,MutS homolog, cyclin-dependent kinase 1 and asparagine synthetase, were expressed in HCC tissue, and formed a single protein signature that predicted HCC survival.Most studies have used clinical proteomic databases including Clinical Proteomic Tumor Analysis Consortium (CPTAC) and Cancer Proteome Atlas (TCPA) as the source for genetic data collection[66].
Forming a gene coexpression network
A research group used 389 differentially expressed genes (retrieved from the GEO database) to build a gene coexpression network using the Robust Rank Aggregation method to aggregate ranked genes, and found that 40 hub genes (i.e., functionally significant in the module formed) were linked to HCC diagnosis, including 30 hub genes that were linked to HCC prognosis. Subsequent clinical validation of those most significant (only three novel biomarkers), was done on 32 HCC patients, showing upregulation in all three biomarkers, and their upregulation was associated with advanced tumor staging and worse prognosis. Those three novel biomarkers had not been assessed before in HCC, and all were linked to the regulation of cellular methylation process[67].
Bioinformatic analysis for HCC-therapy drug candidates (through molecular pathways or drug docking)
An important area in the discovery of novel drug candidates is drug-docking analysis.This is the first line of drug discovery in the era of bioinformatics, and has provided us with research on new applications of existing drugs and discovery of novel ones. This area, despite being interesting, is strictly used by pharmacology and biology specialists, and it is only during clinical validation that clinicians become aware of it during assessment of new drugs in clinical trials.
Followingin silicovalidation, the first step isin vitrovalidation on hepatocellular culture, and laterin vivothrough animal or preclinical trials.
The first human trial is considered Phase 0 and is conducted only on healthy humans, as an exploratory phase prior to examining the treatment on affected patients.Later in Phase I/II, we establish primarily the safety and secondarily the efficacy,while Phase III concentrates mainly on establishing the efficacy of the drug. Finally,the post-marketing phase (Phase IV) determines the effectiveness of the drug in real life settings. Both Phase III and IV also ascertain the occurrence of adverse events (i.e.,safety) in real life settings[68]. In case of known and established drugs already in use for other illnesses, drugs discovered through molecular docking can bypass the animal trials and Phase 0, and go straight to phase I/II clinical trials (Figure 5).
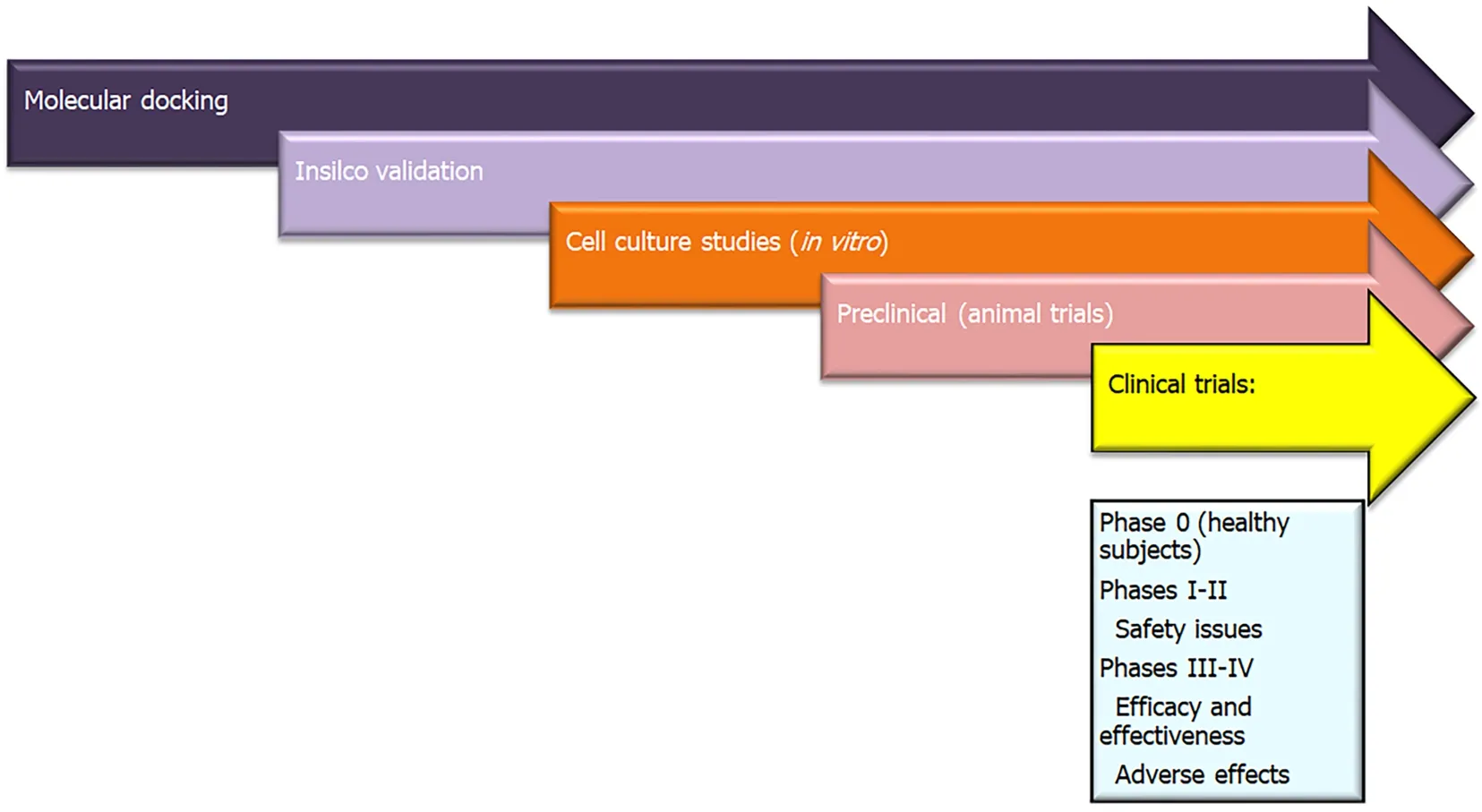
Figure 5 Role of molecular docking from drug discovery to clinical use.
In HCC, many studies considered molecular docking as a way to discover new drug targets. Different pharmacological compounds were considered as drug targets, for example, berberine, which affects the PI3K/AKT signaling pathway[69], or phytoconstituents ofCocculus hirsutus(coclaurine, haiderine and lirioresinol), which affects the VEGF receptor pathway[70]. A recent study used both molecular docking and genetic networks to design anti-HCC drugs from an ancient traditional Chinese medicine SiNiSan (SNS). SNS affects primarily the p53 pathway, thus regulating apoptosis[71].
Drug docking requires knowledge and experience in computational programs and algorithms. An easier and more approachable way to search for drug candidates is through selecting pharmacogenic compounds achieved by bioinformatic analysis of different molecular pathways, then proceed toin vitroanalysis in cellular culture,followed byin vivoanalysis in animal trials, then all through the aforementioned steps of validation. Our team has just published a paper on this topic, where cyan was used as an antioxidant for the inhibition of HCC proliferation, through modulation of the cell cycle in Wistar rats. The drug effect resulted in lower levels of expression of long noncoding RNA MALAT1, and tubulin 1 mRNA, and higher levels of expression of miR-125b. We chose this drug target througha prioribioinformatic analysis, followed by laboratory confirmation, and later byin vivoanimal trials[72].
Other areas of bioinformatics research include whole-exome sequencing, transcriptome sequencing, and cistrome analysis[73].
LlMlTATlONS TO USlNG IN SILICO ANALYSlS AND CLlNlCAL VALl-DATlON
Cost-effectiveness
Data mining is an option to examine the association between genetic material and clinical diseases. Meanwhile, the data collection cost is high. Moreover, most genetic and epigenetic biomarkers incur a high cost for laboratory assessment, mostly supplemented by grants or national or international funding.
Cost-effectiveness of applying those novel biomarkers for general clinical practice has yet to be determined. This needs large-scale population studies, and specifically designed cost-effective models[74-76].
Generally speaking, a novel biomarker should be cost-effective to be applicable in clinical practice guidelines after its wide-scale validation. Ultrasound has proven to be cost-effective in screening, with or without the addition of AFP, as a part of the twostage biomarker–ultrasound screening[77]. This is a critical issue, not only in developing and low-income countries but also in developed countries, while designing their clinical guidelines by healthcare system authorities.
Another problem faced is that laboratory analyses mostly require highly specialized researchers to handle the genetically fragile materials efficiently and without contamination or destruction. Preferably, this kind of research is conducted in highly specialized research laboratories for genetic analysis. Bioinformatics laboratories should always be constructed as a part of these physical laboratories.
CONCLUSlON
The future holds out hope for personalized medicine, where we can treat HCC on an individual level, through assessing the genetic and epigenetic background of the patient, and then planning a specified management, considering the highest benefit and the lowest risk to the patient. The future also offers the promise of early detection of HCC, which has been the main obstacle in achieving our goal of cure, as most of the cases are diagnosed beyond the reach of curative methods, in late clinical stages.
Moreover, we can offer the chance for prognostic prediction of overall survival and tumor recurrence in HCC patients.
Proteomics, genomic, epigenomic and transcriptomic analyses provide massive data on the expression profiles of HCC; however, we are still unclear of their exact role or underlying mechanisms of action. Future studies are needed to integrate these data to provide a clear picture of the disease[66]. For example, S100A9 and granulin protein affect the progression of tumor and metastasis[78], and the inclusion complex of curcumin/β-cyclodextrin polymer prohibitsgrowth of HepG2 cell line[79]. These examples provide diagnostic and prognostic biomarkers for HCC severity and clinical progression, and further research on the affected molecular pathways as possible therapeutic targets specific to each patient, i.e., precision medicine[80,81].
Personalized medicine and individual planning for the management of patients with HCC are the future of medicine. To achieve this we need a multidisciplinary team of hepatologists, oncologists, clinical pharmacists, hepatic surgeons, interventional radiologists, nursing teams, psychiatrists, and social workers. All this should take place in specialized facilities, such as tertiary or specialized hospitals, which deal with these special types of cases. These facilities must include data storage access to a genetic bank, a blood and tissue bank, along with the required bioinformatics specialists to enter, retrieve and analyze data when needed. Finally, supportive teams of social workers, supporting family members and friends, while having effective communication with the medical team, are all essential in procuring the best possible outcome for the patient.
