
新生儿惊厥智能诊疗系统真实场景临床实施效果综合评价的多中心临床研究方案
2022-02-24 11:38:56肖甜甜窦亚兰庄德义胡旭红康文清郭琳赵晓芬张鹏严恺严卫丽程国强周文浩
中国当代儿科杂志 2022年2期
肖甜甜 窦亚兰 庄德义 胡旭红 康文清 郭琳 赵晓芬张鹏 严恺 严卫丽 程国强 周文浩
(1.国家儿童医学中心复旦大学附属儿科医院新生儿科,上海 201101;2.国家儿童医学中心复旦大学附属儿科医院临床流行病学研究室和临床试验中心,上海 201012;3.厦门市儿童医院新生儿科,福建厦门 361006;4.成都市妇女儿童中心医院新生儿科,四川成都 610000;5.河南省儿童医院新生儿科,河南郑州 450018;6.西南医科大学附属医院新生儿科,四川泸州 646099;7.昆明市儿童医院新生儿科,云南昆明 650103)
1 研究背景
新生儿惊厥是临床中最常见且急需诊治的危重症之一。在新生儿期,惊厥往往是中枢神经系统疾病首发且唯一的临床表现,其发生率在不同胎龄、体重和日龄新生儿中的异质性大。总体来说,惊厥最常发生于出生后第1周,发生率为1.5/1 000~5.5/1 000,在新生儿重症监护病房的发生率在8.6/1 000。一项基于连续脑电图(continue electroen-cephalography,cEEG)确诊的新生儿惊厥队列研究发现,胎龄31+0~36+6周新生儿惊厥的发生率为5.0/1 000,28+0~30+6周为54.9/1 000,小于28周为85.6/1 000[1]。常见的病因主要包括新生儿脑病、缺血性和出血性脑卒中、全身感染或中枢系统感染、代谢异常、遗传相关因素。研究表明,惊厥病因、发作频率和时长及抗惊厥药物的使用均与神经系统的远期发育密切相关[2-3]。因此,及时准确的诊断,明确病因,进行正确的抗惊厥药物治疗及对因治疗,可进一步减少或逆转脑损伤[3]。但与其他年龄阶段相比,新生儿惊厥有其特殊性[4-5]。比如,电-临床相关性差,2/3以上的惊厥发作为亚临床惊厥;大部分为局灶性发作;部分患儿在抗惊厥药物使用后,虽无明显的临床发作形式,但可见持续的电惊厥发作。因此,美国临床神经生理学会推荐8通道cEEG监测作为新生儿惊厥诊疗的标准方式,且电惊厥发作为新生儿惊厥的诊断金标准[4,6]。
但在临床工作中,由于判读原始cEEG的专科医生有限,因此抗惊厥药物的使用主要依赖临床观察性判断,但惊厥诊断的准确性仅为50%。临床医生难以获取及时的脑电图报告,给新生儿惊厥的诊断治疗带来了难度。近年来,随着机器学习技术的发展,基于原始脑电信号的自动化惊厥报告系统或可弥补临床这一短板,实现新生儿惊厥的精准诊疗。然而,目前在新生儿惊厥领域基于机器学习的相关临床研究存在以下局限性:(1)机器学习技术主要聚焦在惊厥诊断,极少研究探讨其对新生儿惊厥的治疗策略、预后的影响[7];(2)缺乏机器学习技术在惊厥诊断中的泛化性能评价[7];(3)目前模型的训练样本量均较小,约在6~56个样本不等[8];(4)构建模型的对象均为足月儿,且病因单一,几乎均为新生儿缺氧缺血性脑病[7,9]。然而在真实的临床场景中,早产儿为高发人群且导致惊厥的原因复杂多样;(5)基于真实新生儿神经重症场景中的应用研究极少[9]。
因此,本研究的目的是基于前期开发的适用于不同胎龄、不同病因的新生儿惊厥智能诊疗系统,拟在全国6家新生儿重症监护单元实施惊厥智能诊疗系统对惊厥高危儿的诊断价值评价及临床疗效、预后影响的研究。
2 研究目的
通过在多中心进行新生儿惊厥智能诊疗系统的应用,明确在惊厥高危儿中新生儿惊厥智能诊疗系统在真实临床场景中的诊断价值。同时,明确在确诊惊厥高危儿中该智能诊疗系统对抗惊厥药物的使用及对惊厥高危儿神经系统预后的作用。
3 研究内容
3.1 研究设计
根据研究目的,本研究包含两部分研究。第一部分为新生儿惊厥智能诊疗系统的诊断准确性研究;第二部分为新生儿惊厥智能诊疗系统对惊厥高危儿临床疗效和预后作用的随机对照试验。
3.2 研究对象
(1)研究中心:在全国范围内选择三级以上的新生儿重症监护单元[10],同时具备1年以上新生儿振幅整合脑电图(amplitude electroencephalography,aEEG)、8导联原始脑电图的监测和解读经验。
(2)研究对象:临床疑似惊厥或有异常动作的高危患儿,且足月儿发生年龄小于28 d或早产儿校正胎龄不超过40周(未进行脑电图确诊)。具体纳入标准(满足以下条件之一)包括但不限于以下疾病:①诊断或疑似新生儿缺氧缺血性脑病;②头颅MRI或B超提示颅脑结构畸形;③脑梗死;④诊断Ⅱ级以上颅内出血;⑤中枢神经系统感染;⑥诊断或疑似急性胆红素脑病或因严重高胆红素血症需要换血治疗;⑦其他可引起惊厥的疾病:比如严重电解质紊乱或先天遗传代谢性疾病。排除标准:因头颅血肿、头皮破损等不能行连续脑电图监测。
(3)知情同意:研究前,由研究人员向研究对象监护人详细讲解本研究的目的和内容,使其充分知情研究内容,研究对象的监护人自愿签署知情同意书后方可开始试验。
4 研究方案
4.1 新生儿惊厥智能诊疗系统的诊断准确性研究
4.1.1 数据收集及检测方法惊厥发生诊断的数据采集:研究对象住院期间,采用cEEG(新生儿脑电测量系统OS-610)按照新生儿临床脑电图操作规范采集脑电信号[6]。每次脑电图监测期间至少出现1次临床疑似惊厥异常动作,若监测6 h以上仍未出现临床疑似惊厥异常动作,可根据临床需求停止监测。先由经培训后的固定医师采用新生儿惊厥智能诊疗系统,诊断研究对象的惊厥发生情况。完成cEEG监测后7 d内,对负责cEEG结果读取的医师设盲,令其在完全不知晓智能诊疗系统诊断结果的情况下,读取cEEG结果并进行惊厥发生的诊断。
待测标准:新生儿惊厥智能诊疗系统。诊断方法:研究对象住院期间,由固定医师采集研究对象的cEEG直接上传至电脑端的新生儿智能诊疗模块(包括原始脑电信号量化处理模块,量化脑电信号特征分析和特征提取模块,输出诊断结果)。根据智能诊疗系统的输出结果(是否惊厥),明确研究对象的惊厥发生诊断。
金标准:cEEG结果诊断。诊断方法:研究对象完成智能诊疗系统诊断后的7 d内,对3名专业培训合格、cEEG结果读取经验丰富的医师设盲,令其在完全不知晓模型诊断结果的情况下独立读图,评估诊断研究对象是否发生惊厥。先由2名医生对cEEG进行解读,如2名医生的诊断不同,则由第3名医生进行最后判读。新生儿惊厥发生的诊断标准:在脑电图上发现逐渐演变(即异常EEG波形形态改变或位置跨头部区域转移)的明确异常的cEEG模式,振幅>2μV且持续时间≥10 s[11]。
4.1.2 样本量计算本研究的样本量基于智能诊疗模型诊断惊厥发生的曲线下面积(areas under the curves,AUC)进行计算。既往研究报道的智能诊疗系统诊断高危新生儿惊厥发生的AUC为0.95[11],课题组前期研究数据预计本研究的新生儿惊厥智能诊疗模型可达到的AUC为0.915。进一步结合本研究人群临床特征,应用PASS16.0软件“Tests for One ROCCurve”模块计算,假设本研究人群中智能诊疗模型诊断惊厥发生的AUC与既往研究相差0.035,取α=0.05,Power=0.90,至少需要经金标准cEEG诊断为惊厥发生、无惊厥发生的患儿各167名。考虑10%脱失率,最终确定需要金标准cEEG诊断为惊厥发生、无惊厥发生的患儿各185名。
4.1.3 统计学分析符合正态分布或近似正态分布的连续性变量以均数和标准差进行描述,偏态分布的连续性变量以中位数及四分位数间距描述,以绝对数(百分比)描述计数资料。应用受试者工作特征曲线获得AUC及95%置信区间(confidence intervals,CI),获得新生儿惊厥智能诊疗模型诊断惊厥发生的灵敏度和特异度。统计分析采用Stata 16.0软件(Stata Corp,Texas,USA)进行,以双侧检验P<0.05为差异有统计学意义。
4.2 新生儿惊厥智能诊疗系统对惊厥高危儿临床疗效和预后作用的随机对照试验
4.2.1 基线资料收集符合纳排标准的研究对象在签署知情同意书被纳入研究后,根据制定的病历登记系统,收集相关基本临床信息包括人口学信息、临床判断首次惊厥发生的时间、临床诊断、抗惊厥药物使用时间及剂量等。
4.2.2 随机化分组本研究采用中央区组随机化方法,每4名研究对象为一个区组,按1∶1的比例被随机分配到智能诊疗系统辅助组或常规临床诊疗组。随机化方案由复旦大学附属儿科医院临床试验中心的独立统计团队在SASversion 9.4软件中完成。按照随机分配序列确定的干预方案将由统计团队放置在按区组依序编号、非透明的密封信封中,具体为每个区组的4个分组方案分别被依序放置在编号1~4的4个非透明、密封的小信封中,再统一放入同一个标有相应区组编号的大信封中,交由牵头中心复旦大学附属儿科医院的1名不参与实施干预或者结局观察的中心协调员管理。各分中心在纳入每个区组的第1个研究对象之前,由分中心协调员与牵头中心协调员联络,牵头中心协调员将标有区组编号的大信封按联络先后顺序分配至各分中心。各分中心医生评估患者的合格性、获得其监护人知情同意后将其纳入研究,随后与分中心协调员联系获取该患者的分配方案。分中心协调员依照签署知情同意书的顺序,严格按照信封编号顺序依次拆开信封,然后向参与研究的医生告知研究对象的分配方案,并填写书面登记表。研究对象和实施研究的医生均知晓研究对象的分组,负责结局评估的医生和数据统计分析团队完全不知晓分组情况。
4.2.3 干预措施及临床决策对照组:研究对象入组后,采用cEEG仪,按照新生儿临床脑电图操作规范安置脑电电极采集脑电信号(图1)。该仪器可同步显示aEEG及cEEG。在监测过程中,惊厥的诊断参照临床表现(如典型的强直动作)、aEEG、cEEG综合信息做出判断。对于抗惊厥药物的适应证和使用说明参照图2。对于aEEG和cEEG的结果解读根据各中心诊疗常规进行。
试验组:研究对象入组后,采用和对照组完全相同的方法和仪器采集脑电信号(图1)。在常规监测过程的基础上,辅助连接惊厥智能诊疗系统,该系统可每15 min自动分析1次之前记录的cEEG数据,随后报告患儿是否发生电惊厥,阳性结果会通过报警系统进行床旁提示。临床医生在接受到报警信号后,根据智能诊疗系统的报告结果查看aEEG和cEEG的情况,综合临床信息做出是否惊厥发生的判断。结果判断和临床决策均需在获得金标准cEEG诊断报告之前进行。对于抗惊厥药物的适应证和使用说明参照图2。对于aEEG和cEEG的结果解读根据各中心诊疗常规进行,与对照组一致。
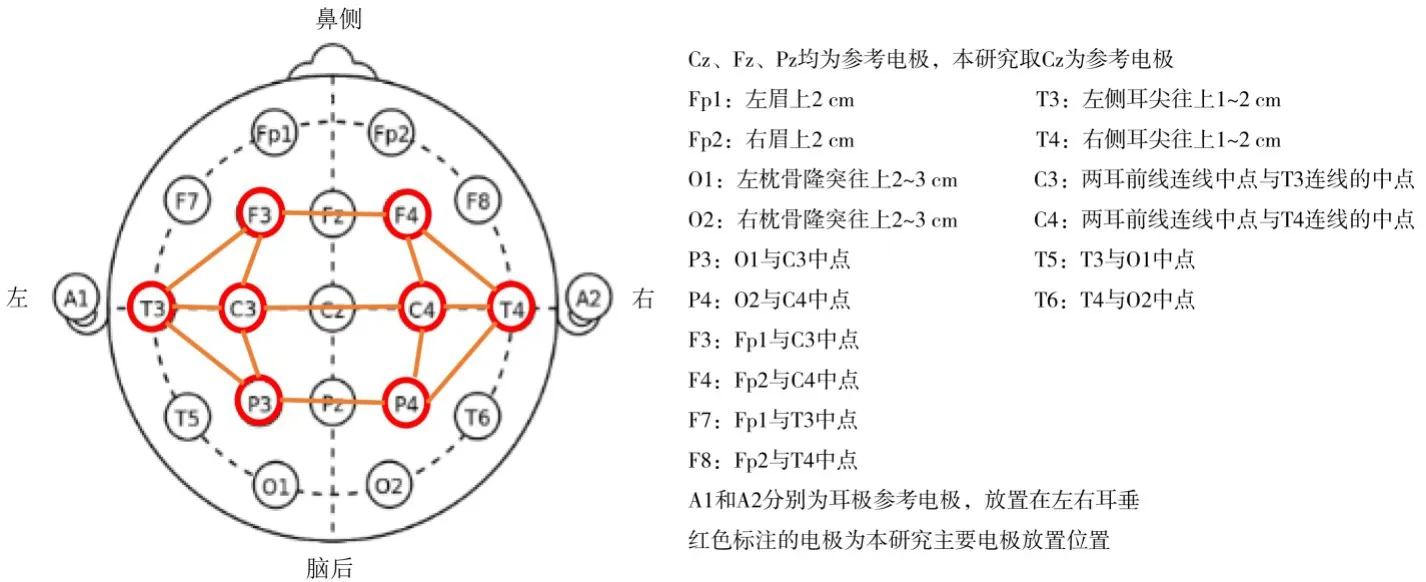
图1 新生儿脑电图采集的标准脑电电极放置位置

图2 新生儿抗惊厥药物使用流程 红色虚线以下表示根据各中心的实际情况选择用药或者会诊。#示维生素B6的使用适应证为除外症状性惊厥(缺氧缺血性脑病、颅内出血、脑结构畸形、中枢神经系统感染、电解质紊乱、低血糖、急性胆红素脑病)。
4.2.4 观察终点首要观察终点为研究对象住院期间cEEG监测结束的时间点,次要观察终点为研究对象纠正胎龄6个月时。
4.2.5 结局指标主要结局指标:抗惊厥药物使用错误率。抗惊厥药物错误使用的定义(满足以下条件之一):经金标准cEEG诊断有惊厥发生,但未用药治疗;经金标准cEEG诊断有惊厥发生,但延迟到正式脑电图报告生成后方才用药;经金标准cEEG诊断无惊厥发生,但使用了抗惊厥药物。
次要结局指标:检测期间电惊厥持续时间、电惊厥负荷、纠正胎龄6个月时Gesell评分等。电惊厥负荷=电惊厥发作次数/监测时间;Gesell发育量表主要评价中枢神经系统的功能,是诊断性量表。该发育评估由康复科医师完成。
结局指标资料收集:根据制定的病历登记系统,填写相关内容。
4.2.6 样本量估计本研究中试验组为智能诊疗系统辅助诊断组,对照组为常规人工诊疗组。样本量计算基于主要结局指标:惊厥高危患儿cEEG监测期间的抗惊厥药物使用错误率。根据课题前期临床病例数据及既往研究[2],预计常规诊疗组的抗惊厥药物使用错误率至少为30%,本研究预期智能诊疗系统的使用能够降低15%的错误率。应用PASS16.0软件进行样本量计算,取Power=0.90,双侧α=0.05,则每组至少各需要161名惊厥高危患儿。考虑10%脱失率,则两组至少各需179例,最终确定该试验两组一共需要至少360例符合条件的惊厥高危患儿。
4.2.7 数据处理数据收集和管理:根据研究方案和数据分析方案,制定病历登记系统并进行论证。为保证关键数据完整性,设定必填项。并事先制定病历登记系统填写和质量保证的标准方法。由事先指定和培训合格的研究者填写,每个入选病例必须完成病历登记系统。由研究者审查并交由数据管理员进行数据录入与管理。
数据录入与管理由专人负责。为保证数据的准确性,应由两个调查员独立进行双次录入,经软件核对及人工纠错后进行核查。中央数据库管理员确认数据库准确无误后,进行数据库锁定。锁定后的数据文件提交给独立统计团队进行分析。
4.2.8 统计学分析研究统计分析由独立的专业统计分析团队承担。根据关键科学问题、明确设计方案,依据首要结局指标属性决定统计分析方法,根据预期效应计算样本量。在研究开始纳入后至最后1例患者数据采集完成期间,进行详细统计分析计划的撰写和论证,上传至临床试验注册网。以下为统计分析策略和方法概述。
本研究中,统计分析均采用意向性分析(intention-to-treat,ITT)策略,仅敏感性分析采用符合方案集分析(per-protocol,PP)。ITT包含所有随机化后的受试者,按原分配方案进行分组分析;PP仅包含符合以下条件受试者:实际中按照被分配方案完成了整个试验,依从性良好。研究对象偏离干预措施的情况包括:入组患儿未及时连接智能诊疗系统,医生在查看智能诊疗结果后未做出治疗或不治疗的决策。
主要结局数据分析:主要结局指标为惊厥高危患儿监测期间抗惊厥药物使用错误率。应用广义线性模型(generalized linear model,GLM)比较两组之间患儿抗惊厥药物使用错误率的差异,将分组作为固定效应,连接函数为binomial distribution和identity link function,获得两组之间的差异效应值(risk ratioor risk difference)及其95%CI。
敏感性分析:采用和上述ITT策略相同的统计学方法,在PP数据集中应用GLM,获得两组之间的差异效应值及其95%CI。此外,在PP数据集中应用GLM,调整协变量后获得差异效应值及其95%CI。
亚组分析:按照胎龄将研究对象分为两层,在每个亚组有足够研究对象的前提下,应用GLM进行主要结局指标的亚组分析。
次要结局数据分析:①连续型变量:符合或近似正态分布的连续型变量应用均数和标准差来描述其离散趋势,非正态分布的连续型变量采用中位数和四分位数间距描述,应用GLM(连接函数为normal distribution和identity link function)获得两组之间的效应值差异及其95%CI。②分类变量:以绝对数(百分比)描述,应用GLM(连接函数为binomial distribution和identity link function)获得两组之间的差异效应值及其95%CI;应用GLM(连接函数为binomial distribution和logit link function)获得两组之间的oddsratios及其95%CI。
缺失数据处理:缺失数据将显示在病历登记系统记录中。在进行数据填补时,根据不同的实际缺失原因采用不同的方法。对于随机缺失的生命体征、实验室指标检测、辅助检查等数据,采用多重填补方法等进行缺失数据填补。
采用Stata 16.0软件(Stata Corp,Texas,USA)或SAS 9.4软件[Copyright(c)2016 by SASInstitute Inc.,Cary,NC,USA)进行统计学分析,所有统计学检验均为双侧检验,设检验水准α=0.05,P<0.05为差异有统计学意义。
5 临床试验伦理、临床研究注册及知情同意
研究者有责任确保本次试验的进行符合中国临床试验质量管理规范及赫尔辛基宣言(经东京、威尼斯、香港、南非、爱丁堡修改的版本)的要求。研究者应确保本次试验由有资格的伦理委员会审阅及批准。
本项目已在美国临床试验数据库(Clinical-Trials.gov)注册(NCT04991779和NCT05036395)。本研究方案已经获得复旦大学附属儿科医院伦理委员会审批[复儿伦审(2021)305]。
在试验开始之前,研究者应将本次试验方案、知情同意书及其他必需的材料交与伦理委员会供其审阅及批准。入选本研究之前,将向受试者充分告知,受试者签署知情同意书后方可纳入研究。
6 安全性评价
研究对象在新生儿重症监护室,因此,若在干预期间发生脑电检测处的皮肤损伤等情况,会尽早告知家长,并详细记录。发现不良反应时,研究者可根据病情采取必要的处理措施,如调整剂量、暂时中断用药等,并决定是否终止试验。出现严重不良事件,承担试验研究的单位必须立即采取必要的处理措施,保护受试者的安全。
7 讨论
本研究方案是基于前期开发的新生儿惊厥智能诊疗系统,拟在真实世界中探索该智能系统的实施效果,最终目的在于解决临床对于新生儿惊厥实时诊疗的瓶颈。与目前现有的惊厥智能系统相比[7,9],本智能系统的优势在于适用于早产儿和足月儿,同时不局限于缺氧缺血性脑病所致的惊厥,可适用于多种病因甚至是遗传病因所致的惊厥。因此,拓展了惊厥智能系统在复杂的临床场景中的应用范围。
目前的共识指出新生儿期原始脑电图的记录时间依据不同的监测目的有所不同[12]。对于高危新生儿脑电图背景的评估,一般采用短程记录,即记录时间一般在2~4 h之间,应该包括至少1个完整的清醒-活动睡眠-安静睡眠周期;对于新生儿惊厥的监测,推荐长程记录,即纪录时间在4 h以上;对于已确诊的惊厥患儿建议监测持续到惊厥停止后的12~24 h。本研究设计中,对于原始脑电图的记录时间的制定除了考虑上述共识外,还考虑了临床的可实际操作性及过长时间监测造成电极对头皮的损伤,因此,规定每次脑电图监测期间至少出现1次临床疑似惊厥异常动作,若监测6 h以上仍未出现临床疑似惊厥异常动作可根据临床需求停止监测。
本研究方案的第2部分涉及到对研究对象抗惊厥药物的使用问题,因此,在设计该研究方案时也充分考虑到常规组中患儿的安全性问题。对于常规组采用了标准常规诊疗方案,即对高危患儿采用脑电图监测,根据aEEG进行观察,结合临床表现,决定用药。抗惊厥用药的标准参考国内外指南[13-15]和本研究专家组共同制定。对于所有研究对象的诊疗操作规范是符合当前诊疗标准规范的,对干预组和对照组患者的临床治疗和预后并不会造成额外的风险和损害。另一方面,我们通过文献复习及前期数据总结预计常规诊疗组的抗惊厥药物使用错误率至少为30%左右[2],这样的临床现状是不容忽视的。因此,本研究旨在探讨通过智能诊疗系统辅助临床判断是否能优化惊厥高危患儿的临床用药现状,以期为该人群的临床诊疗提供进一步建议。
猜你喜欢
睿士(2023年2期)2023-03-02 02:01:09
家庭医学(下半月)(2019年9期)2019-10-12 08:04:06
家庭医学(下半月)(2019年8期)2019-09-25 09:02:00
意林(2018年3期)2018-03-02 15:17:24
妈妈宝宝(2017年3期)2017-02-21 01:22:12
厦门理工学院学报(2016年1期)2016-12-01 04:50:48
现代电生理学杂志(2016年1期)2016-07-10 10:20:58
北京信息科技大学学报(自然科学版)(2016年6期)2016-02-27 06:31:48
川北医学院学报(2015年5期)2015-12-05 08:22:33
现代电生理学杂志(2015年1期)2015-07-18 11:02:17
