
一种基于加权非负最小二乘的蛋白质定量方法
2022-02-23 07:08方言郑浩然
北京生物医学工程 2022年1期
方言 郑浩然
0 引言
液相色谱-串联质谱(liquid chromatography-tandem mass spectrometry,LC-MS/MS)技术是一项高效的蛋白质检测分析技术[1],有效结合了色谱分离复杂样品的能力和质谱的高灵敏度及定性定量能力,广泛应用于“自下而上”(bottom up)的蛋白质定量流程中[2-3]。在此流程中,样品中的蛋白质会被蛋白酶消化成肽段,并依次进行色谱分离、离子化和质量分析,得到质谱数据。通过分析质谱数据能够提取肽段的相对丰度,进而对相应蛋白质进行测定。蛋白质定量作为整个流程的下游,需要尽量减少由上游处理过程带来的累积误差对最终结果的影响(例如样品制备和处理中的错误、质谱仪采集信号的系统误差和噪声干扰、肽段的错误鉴定等)[4]。高效准确地基于已有肽段丰度信息进行蛋白质定量分析是蛋白质组学的一个重要方向,在蛋白质作用机制和生物标志物发现与检测等研究中具有广泛应用场景。
基于质谱的蛋白质组学方法已经成为诊断、预后和治疗性蛋白质生物标志物发现的首选策略,然而由于许多生物标志物以非常低的浓度存在于生物样本中,在实验中通常具有难以检测、可重复性差、准确度低的特点,对蛋白质定量工作提出了挑战[5-7]。目前已经有多种方法能够基于已有的肽段丰度信息进行蛋白质定量分析,常用的方法按照参考肽段(即用于定量的肽段)的规模可以分为三类:(1) 基准型,即从来源于同一蛋白质的所有肽段中选取一个肽段作为基准肽段,以其丰度为基准对其他肽段的丰度进行标准化,从而对蛋白质进行测定。DAnTE[8]选取缺失值最少的肽段作为基准肽段,将各个肽段与基准肽段的丰度比作为其标准化后的丰度,蛋白质丰度为所有标准化肽段丰度的平均数。(2) 采样型,即通过一定策略选取来源于同一蛋白质的部分肽段,将其相对丰度进行累加或均值等计算得到相应蛋白质的丰度。T3PQ[9]认为每种蛋白质的三个离子化效果最佳从而具有最高信号强度的肽段与相应蛋白质的丰度直接相关,并据此以这些肽段的平均丰度来量化相应蛋白质。(3) 参数型,即结合已有的蛋白质与肽段信息,以蛋白质丰度作为参数进行建模,通过评估参数进行蛋白质定量分析。all-P[10]假定肽段的强度服从对数正态分布,在无信息先验分布的贝叶斯分层框架中构建非线性模型,从而对蛋白质丰度进行估计。
由于选取策略灵活且实现相对简单,基准型方法和采样型方法被应用于各种定量蛋白质组学实验中,然而它们通常要求所选取的肽段是高丰度肽段或唯一肽段,没有充分利用其他肽段信息,在对低丰度蛋白质的准确定量方面具有局限性。此外,在大规模蛋白质测定实验中,由于样品的高度复杂性,不同蛋白质之间可能存在众多同源性序列[11],导致某些肽段来自多种不同的蛋白质,难以确定各个来源的贡献,通常在蛋白质定量时被忽略,因此往往存在蛋白质没有足够参考肽段可用的情形[12-13]。参数型方法则不受此类肽段约束,通过分析更多的肽段信息进行更全面的蛋白质丰度估计,尽管会导致蛋白质定量问题更加复杂,并且可能引入新的误差,但若模型设计得当,也为达到更高的定量准确度提供了潜在可能性[14]。另一方面,目前蛋白质定量方法普遍将参考肽段同等看待,而实际上由于各种性质差异,不同肽段对蛋白质定量的影响也有所不同。为了准确检测低丰度蛋白质,需要充分利用所有肽段的信息,并对不同肽段的重要性加以区分,较为合理的方法是对各肽段赋予不同权重。
本文提出一种参数型方法WeQuant,利用无标记蛋白质组学实验中的肽段质谱分析结果构建加权非负最小二乘模型,进而实现对不同浓度范围的蛋白质准确定量。
1 WeQuant算法
1.1 构建肽段-蛋白质关系矩阵
关系矩阵是一种反映肽段与蛋白质关系的常用手段。蛋白质通过蛋白酶的作用水解为若干质量较小的肽段,而不同的蛋白酶作用于蛋白质的不同位点,因此酶解产生的肽段也有所差异。目前实验中应用最多的酶为胰蛋白酶[15]。基于实验使用的特定蛋白酶,可以推导出蛋白质酶解产生的肽段集合,构建蛋白质酶解向量。在一个蛋白质的酶解向量中,使用1和0分别标记该蛋白质与一个肽段是否存在酶解关系。将所有蛋白质酶解向量组合得到的矩阵即为蛋白质与肽段的关系矩阵。
1.2 加权非负最小二乘模型
假设M∈Rm×n为肽段-蛋白质关系矩阵(m个肽段,n个蛋白质),Q∈Rm×1为测定的m个肽段丰度,P∈Rn×1为估计的n个蛋白质丰度。
WeQuant算法基于以下假设:肽段的丰度是与其存在酶解关系的不同蛋白质的丰度之和[10,16]。目标是找到最能解释Q的蛋白质酶解向量(M各列)的最佳非负线性组合,P被确定为相应的非负最小二乘问题的解:
(1)
s.t.Pj≥0,j=1,…,n
式中:W为权重矩阵;wi(i=1,…,m)为第i个肽段的权重。
在实际情况下,关系矩阵通常是超定的(所鉴定的肽段数量多于蛋白质数量),并且具有线性独立的列(无法将一个蛋白质酶解向量表示为其他蛋白质酶解向量的线性组合),从而具有唯一的全局最小值。本文通过将梯度下降应用于目标函数以确定此全局最小值。
1.3 肽段权重分配
在加权非负最小二乘模型中,权重越高的肽段对目标函数值影响越大。为肽段分配合理权重是决定模型性能的关键。
一方面,从肽段种类考虑,相比于唯一肽段,共享肽段的组成更加复杂,而匹配蛋白质越多的肽段通常其丰度误差越大,因此其权重应越低。与肽段相匹配的蛋白质数量能够反映肽段组成的复杂程度:
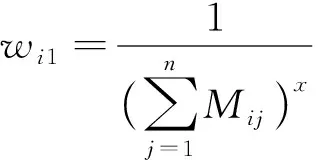
(2)
式中:wi1为反映第i个肽段来源复杂度的特征值;x>0,用于控制匹配不同蛋白质数量的肽段的权重差距。
另一方面,从肽段丰度考虑,通常丰度越高的肽段受到系统噪声的影响越小,因此其权重应越高。由于不同实验得到的肽段相对丰度通常不具有可比性,因此需要进行丰度标准化,使其处于相同数量水平。通过合适的函数f对肽段丰度向量Q进行标准化:
wi2=f(i)
(3)
式中:wi2为第i个肽段的丰度特征值,由标准化函数f确定。
为综合两方面的影响,以肽段的两个特征值为坐标,将肽段映射为坐标轴上一点,并以该点距原点的欧式距离作为其权重:
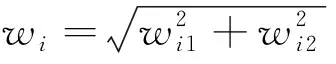
(4)
式中:wi为第i个肽段的权重。对于参数x与f,将通过具体实验分析不同参数设置对模型性能的影响,从而确定合适取值。
2 实验评估
2.1 数据集
LFQbench质谱数据集[17]是为评估定量准确度而专门设计的通用数据,由3个物种的蛋白质酶解物以2种比例(A和B,如表1所示)分别混合后各进行3次重复 SWATH-DIA[18-19]采集得到。采用LFQbench对该数据集进行分析得到的肽段定量数据作为实验数据集。

表1 样品组成Table 1 Sample composition
该数据集含有在上述实验中累积鉴定42 439个肽段的丰度信息,分别来自于6581个蛋白质,其中人、酵母和大肠杆菌(E.coli)的肽段数量及对应蛋白质数量如表2所示。
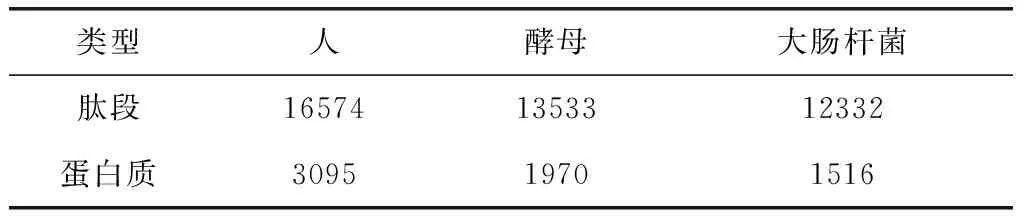
表2 肽段与蛋白质数量分布Table 2 Quantity distribution of peptide and protein
2.2 模型参数选择
为了最大程度提高WeQuant模型的性能,使用不同的参数设置在给定数据集上进行测试,并分析其对模型性能的影响。
对于用于标准化肽段丰度的函数f,分别采用以下3种方法:
(5)
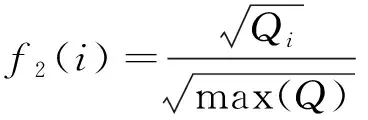
(6)
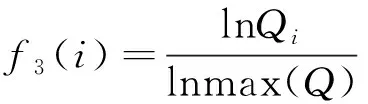
(7)
式中:max为最大值函数;Q为肽段丰度向量。
对于每一种标准化函数f,对参数x分别使用不同取值(x=0.25,0.5,1,2,3,4,5)进行WeQuant定量分析。图1展示了每组参数实验的定量准确度,即3个物种蛋白质的A和B丰度之比log2(RA/RB)的平均中位数绝对偏差。
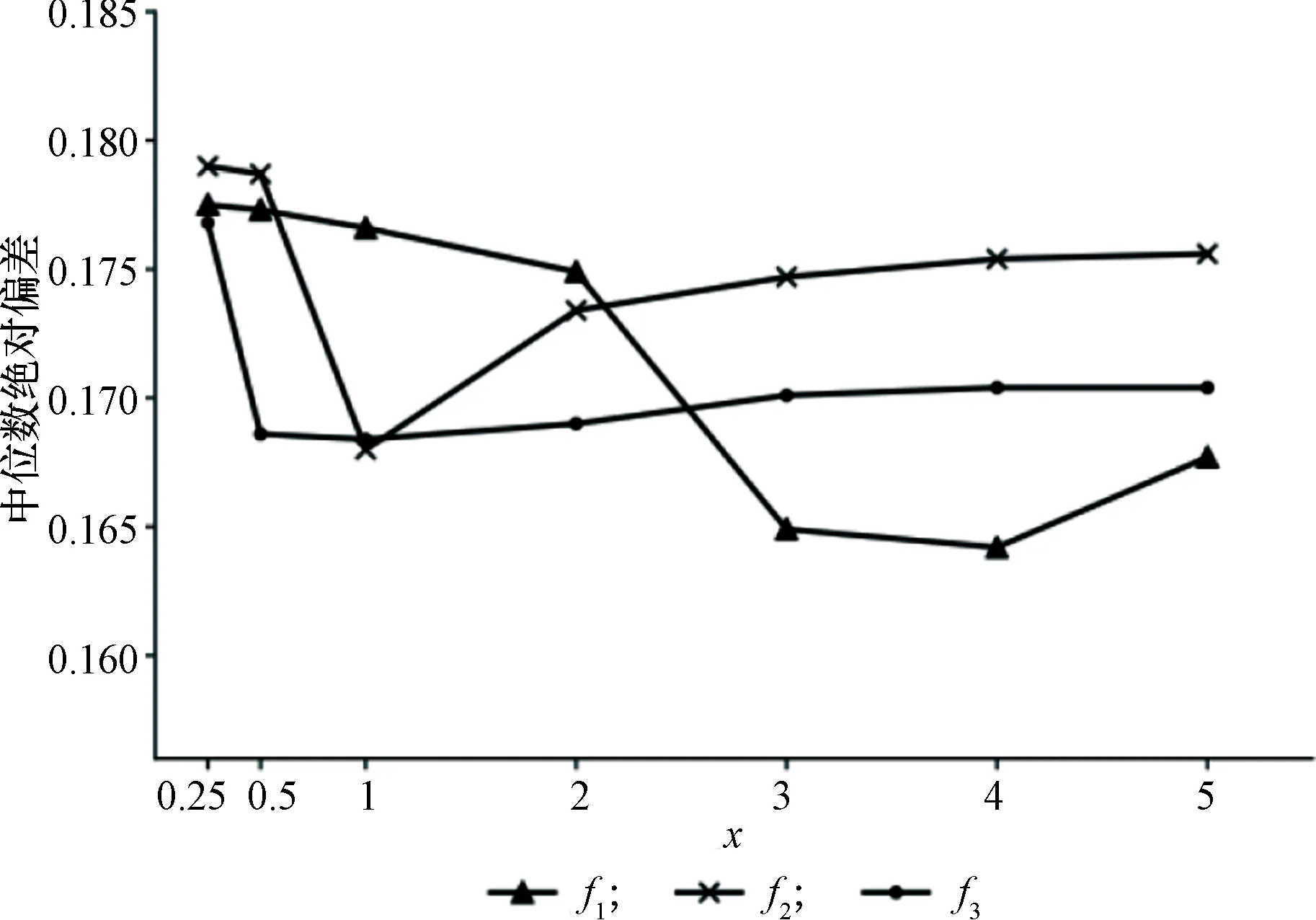
图1 不同参数设置的定量准确度Figure 1 Quantitative accuracy of different parameter settings
在每个特定标准化函数f的作用下,随着指数x的增大,WeQuant算法的定量准确度具有先提高后有所降低的整体趋势。指数x的增大使得唯一肽段和共享肽段(包括匹配不同蛋白质数量的肽段)的权重差距逐渐加大,表明两种肽段对算法性能均有贡献,WeQuant能够有效利用共享肽段信息提高蛋白质定量的准确度。
在标准化函数方面,相比于f3,其他两种标准化函数对x的变化更加敏感。在x值较小的情况下,f2和f3的定量准确度比f1高。随着x的增大,f1能够达到更高的准确度,并且超过f2和f3的最佳准确度。
根据以上实验结果,最终确定WeQuant算法在该数据集上的最佳参数选择,即x=4,f=f1。
2.3 多方法性能对比
为了更全面地评估WeQuant性能,从三类无标记蛋白质定量方法中各选取一种典型方法(DAnTE[8]、T3PQ[9]和all-P[10]),并在LFQbench数据集上对比了它们与WeQuant的性能。
首先,在蛋白质数量方面,WeQuant能够有效定量5 869个蛋白质(即在重复实验中定量2次以上),相比于其他方法平均提高了17%。图2展示了WeQuant有效定量蛋白质的丰度分布,包括WeQuant特有的蛋白质(12.3%)以及和其他方法共有的蛋白质。值得注意的是,WeQuant能够有效定量较低丰度范围的蛋白质,而这些蛋白质未被其他方法有效定量。
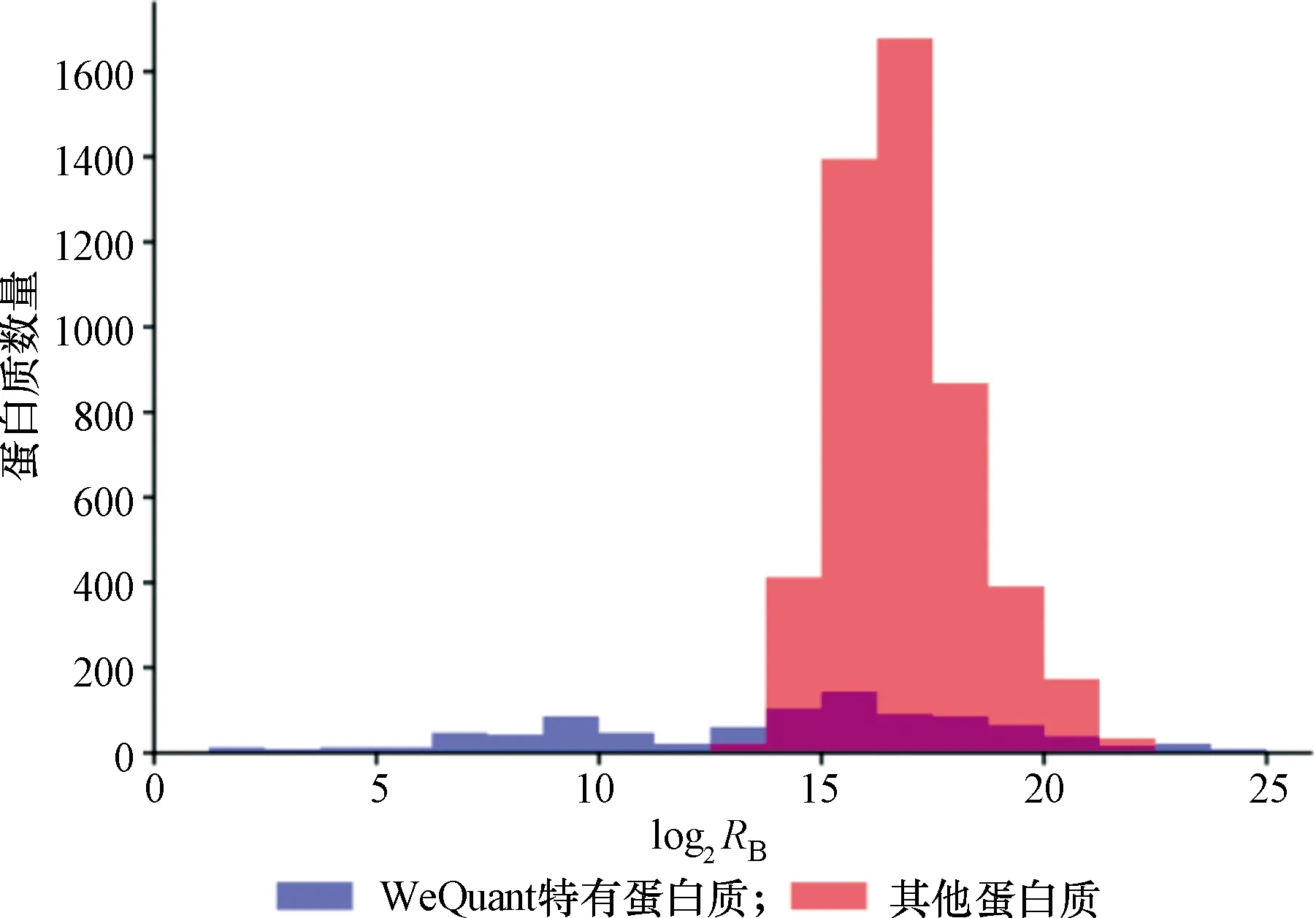
图2 WeQuant定量蛋白质的丰度分布Figure 2 Distribution of protein abundance for WeQuant
然后进一步分析这些方法在各物种上的定量准确度。对于每个方法,将其定量的蛋白质按照物种及其B样品丰度的三分位数进行分组(共计9组),分别统计每组蛋白质的定量准确度,如图3所示。
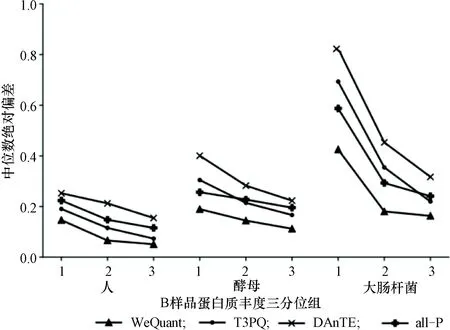
图3 不同方法的各组定量准确度Figure 3 Quantitative accuracy of each group by different methods
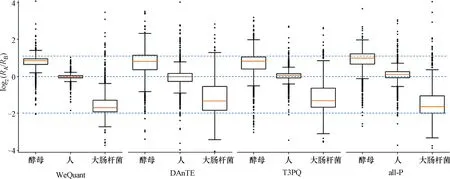
图4 蛋白质在A/B样品之间的相对丰度比值箱线图Figure 4 Box plot of the relative abundance ratios of protein between A/B samples
与其他方法相比,WeQuant在3个物种上均取得了最高的定量准确度。在所有方法中,高丰度蛋白质组的定量准确度更高,并且每个物种均在丰度最高的蛋白质组上达到了最佳效果。基准型方法DAnTE由于参考的肽段过少,定量容易受到噪声影响,特别是在低丰度蛋白质上,其丰度比具有更高的离散程度。采样型方法T3PQ和参数型方法all-P参考了更多的肽段,通常可以减小噪声的影响,而前者更依赖于高丰度的肽段,后者考虑了更多肽段,因此在高丰度蛋白质上T3PQ准确度更高,相应地在低丰度蛋白质上all-P准确度更高。
由各方法分析得到的3个物种蛋白质的实际丰度比与理论丰度比存在不同程度的差异。如图4所示,对于所有方法,人蛋白质的丰度比最接近理论值,酵母和大肠杆菌蛋白质均出现更大程度偏离,而大肠杆菌偏离程度最高。注意到酵母和大肠杆菌在样品中的比重较低,这表明背景消除(background subtraction)的潜在问题导致它们在酵母和大肠杆菌的丰度比上存在系统偏差[17]。此外,相比于其他所有方法,WeQuant在3个物种上均具有最小的偏差。特别是对于大肠杆菌蛋白质,WeQuant较其他方法显著降低了实际丰度比与理论值的差异,表明WeQuant能够有效利用低丰度肽段信息进行蛋白质定量。
3 讨论与结论
作为自下而上的无标记蛋白质定量流程的下游工作,基于已有肽段丰度信息的蛋白质定量分析需要有效降低上游过程误差带来的影响,从而保证最终定量准确可靠。目前的蛋白质定量方法较多地依赖于高丰度肽段或唯一肽段,因此对于低丰度蛋白质的定量准确度仍然有待提高。对于这一问题,WeQuant基于传统蛋白质定量方法的思想,从肽段的丰度和来源两个维度对各肽段进行综合评估,赋予各肽段不同的权重,并以此建立加权非负最小二乘模型,从而实现对不同丰度范围的蛋白质进行准确定量。在实验数据集上的结果表明,相比于已有方法,WeQuant在显著增加有效定量的蛋白质数量同时,进一步提高了蛋白质定量的准确度,特别是在低丰度蛋白质上的性能更加突出。
WeQuant重在提出一个充分利用全部肽段信息进行蛋白质定量的模型,本文采用的权重计算方法并非唯一的,也不排除存在更优的权重计算方法,用户可以结合相关理论自主定义。此外,LFQbench数据集被广泛应用于无标记定量蛋白质组学方法的基准化分析,能够较全面地评估蛋白质定量方法的关键性能指标,特别是对比不同丰度范围蛋白质的定量差异,因此本文仅采用该数据集对提出的蛋白质定量方法进行评估。后续工作将采用更多数据集,并致力于分析肽段的各种因素对蛋白质定量的贡献与影响,设计更加高效的权重计算方法,进一步提高蛋白质定量的各项性能指标。
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09
科海故事博览·下旬刊(2022年4期)2022-05-07
现代仪器与医疗(2022年1期)2022-04-19
现代仪器与医疗(2021年2期)2021-07-21
分析化学(2019年3期)2019-03-30
分析化学(2018年12期)2018-01-22
价值工程(2016年32期)2016-12-20
中学生数理化·高一版(2016年8期)2016-12-07
考试周刊(2016年34期)2016-05-28
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
