
CAPN3基因杂合缺失合并半合子突变致肢带型肌营养不良2A型2例☆
2022-02-22 05:42连飘飘徐岩
中国神经精神疾病杂志 2022年12期
连飘飘 徐岩
肢带型肌营养不良2A型(limb-girdle muscular dystrophy type 2A,LGMD2A)是一种常染色体遗传病,在欧洲为最常见的LGMD类型[1],但在亚洲特别是我国相对较少见。LGMD2A有高度临床异质性,发病年龄、临床表现及疾病的进展速度均有较大变异,诊断难度较大。本文报告了一对临床表现类似Becker型肌营养不良(Becker muscular dystrophy,BMD)的孪生兄弟,通过基因测序发现钙蛋白酶3(calpain 3,CAPN3)基因的复合杂合变异,最终确诊为LGMD2A,显示了基因检测在 LGMD2A患者诊断中的价值,为该病的临床诊断和基因检测提供更多参考依据。
1 临床资料
1.1 临床特征病例1(先证者),男,34岁,因“四肢乏力10余年,加重伴行走不稳、下肢麻木4年”于2020年11月16日入院。患者10年前无明显诱因出现四肢乏力,主要表现为活动易疲劳,休息后逐渐缓解,4年前患者感四肢乏力较前明显加重,下肢乏力为甚,逐渐向上肢发展,伴行走不稳,活动略缓慢,双侧大腿外侧麻木,遂就诊于我院。既往强直性脊柱炎4年,家族中同胞哥哥有类似病史。体格检查:高级神经活动正常,脑神经检查未见异常,无明显肌萎缩,四肢近端肌力5-级、远端5级,肌张力均正常,双侧腓肠肌无肥大,无肌肉压痛,伴有漏斗胸,翼状肩,肱二头肌肌腱反射可,余腱反射减退,膝反射消失,病理反射未引出,直线行走较差,下蹲起立可。入院后完善相关检查: 肌酸激酶(creatine kinase,CK)682 U/L(正常值38~174 U/L),乳酸脱氢酶76 U/L(正常值109~245 U/L),雌二醇222.90 pmol/L (正常值41~159 pmol/L)。其他常规结果均正常。心电图:窦性心律不齐。大腿MRI:双侧大腿肌肉萎缩伴结缔组织脂肪化,以后群、内侧群肌肉受累最为严重,大腿前群肌肉亦有累及,但程度较轻。肌电图提示肌源性损伤,神经传导速度正常。肌肉活检(右股内收肌):肌纤维轻度大小不等,少数肌纤维萎缩,见个别核聚集,未见明显变性坏死肌纤维(见图1)。
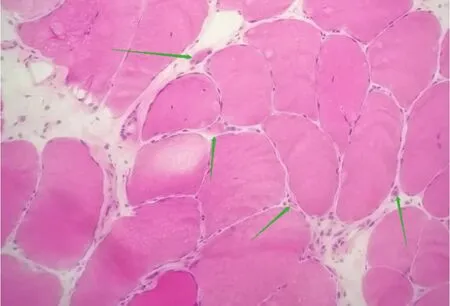
图1 肌肉组织病理检查苏木素-伊红(HE)染色结果 肌纤维轻度大小不等,少数肌纤维萎缩,见个别核聚集,未见明显变性坏死肌纤维、肌裂或核内移,箭头示部分肌纤维萎缩。
病例2(先证者之兄),男,34岁,为例1的同胞哥哥,四肢乏力表现与先证者类似,但程度较轻,既往有先天性白内障,强直性脊柱炎4年。体格检查:高级神经活动正常,脑神经检查未见异常,异常乳房肥大,无明显肌萎缩,双侧小腿肌肉假性肥大,鸭步,四肢近端肌力5-级,远端肌力5级,肌张力正常,双上肢腱反射减退,左下肢膝反射消失,右下肢膝反射减退,双侧跟腱反射存在,病理反射未引出。入院后完善相关检查:CK 567 U/L,乳酸脱氢酶270 U/L,α-羟丁酸脱氢酶186 U/L(正常值72~182 U/L),甘油三酯2.67 mmol/L(正常值<1.7 mmol/L),孕酮:0.51 nmol/L(正常值<0.474 nmol/L),雌二醇:224.9 pmol/L,其他常规检查结果大致正常。心电图:窦性心动过缓伴不齐,完全性右束支传导阻滞。心脏超声:心脏舒张功能减退。大腿MRI:大腿内侧群、后群轻度萎缩伴脂肪化。肌电图提示肌源性损伤,神经传导速度未见明显异常。
1.2 基因检测与结果首先采用高通量测序(highthroughput sequencing,HTS)对先证者(Ⅱ2,见图2)外周血DNA样本进行肌肉病相关基因外显子捕获测序,结果显示CAPN3基因第4号外显子上存在错义突变 c.526G>A(p.Val176Met),第176位的遗传密码子改变,氨基酸从缬氨酸突变为蛋氨酸。c.526G>A曾被文献提及过,在ClinVar数据库中被归类为临床意义未明突变。先证者该变异位点显示为纯合,但外显子测序分析软件提示受检者可能携带了一段位于基因组 15q15.1处约 19.3 kb片段的杂合缺失变异。进一步行Sanger测序验证(见图3),针对4号外显子设计引物序列:正向引物5'- TGTGTGTAAGAAGAACTGGTGG -3',反向引物为5'- CCTGTGGGTGTCTTGGTTAT -3',对目标区域片段进行PCR测序。结果表明,先证者及其胞兄(Ⅱ1)确实存在纯合突变,其父(I1)为c.526G>A(p.Val176Met)变异携带者,但其母(I2)未检测到上述位点变异。最后,qPCR对家系中CAPN3基因的15q15.1区域进行检测(表1),发现先证者、先证者胞兄及母亲,都存在该区域的杂合缺失,验证了外显子测序分析结果,是由于CAPN3基因2-14号外显子杂合缺失使4号外显子点突变处于特殊的“半合子”状态。目前为止,CAPN3基因15q15.1区域杂合缺失未被报告。该变异包含CAPN3基因的第 2~14 号外显子,可能会导致编码的蛋白功能丧失,并在DGV普通人群数据库中未检出该变异,依据美国医学遗传学与基因组学学会 (American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南[2]为疑似致病性变异。只有患者为复合杂合突变,只携带单个点突变或缺失的家系携带者表型未见异常,表明基因突变与表型存在共分离。综上所述,根据ACMG 遗传变异分类标准与指南,该复合杂合突变是致病突变。
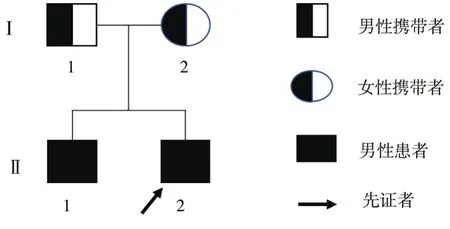
图2 先证者家系图 先证者父亲(I1)为c.526G>A(p.Val176Met)变异携带者,先证者母亲(I2)存在15q15.1(chr15:42676681-42695975)杂合缺失变异,先证者(Ⅱ2)及其孪生哥哥(Ⅱ1)存在c.526G>A(p.Val176Met)和(chr15:42676681-42695975)杂合缺失的复合杂合变异。
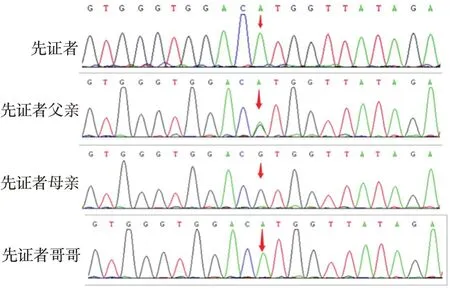
图3 先证者家系sanger测序 Sanger测序验证CAPN3基因的杂合突变c.526G>A(p.Val176Met),箭头表示c.526G>A突变所在位点。

表1 先证者家系CAPN3基因突变的qPCR检测
2 讨论
LGMD2A是CAPN3基因突变引起的常染色体隐性遗传病,在最新的基于遗传模式的命名方式中被称为LGMD R1 calpain3-related[3]。LGMD2A主要临床特征为骨盆和肩部近端肌肉进行性、对称性的无力,多于儿童或成人早期起病,下肢重于上肢,多伴翼状肩及跟腱挛缩,心脏和呼吸系统并发症在 LGMD2A型中并不常见[4]。病程早期血清CK水平明显升高,后期逐渐下降[5]。肌肉影像学以下肢大腿肌肉受累为主,主要是大腿内收肌、后群肌和臀小肌[6]。LGMD2A患者的肌肉活检表现为肌营养不良的特征,分叶状肌纤维是其典型病理改变,但缺乏特异性。本研究中,即使2例患者具有相同的CAPN3基因变异,他们亦有不同的临床表现,先证者临床症状较其兄严重。
临床上,假肥大型肌营养不良(Duchenne/Becker muscular dystrophy,DMD/BMD)可能表现出和LGMD2A相似的肌无力发作,且DMD/BMD更为常见,应注意加以鉴别诊断,尤其例2有双侧小腿肌肉假性肥大的临床表现,这一体征是DMD/BMD的典型临床表现[7]。LGMD2A和BMD均可于青年时期起病,疾病进展缓慢,表现为四肢近端肌肉无力萎缩,肌电图呈肌源性损伤,肌肉活检呈肌纤维变性、坏死,脂肪和结缔组织增生等特点。但两者在一些临床表型和辅助检查中仍存在差异。LGMD2A常伴翼状肩和跟腱挛缩,BMD经典体征是双侧腓肠肌肥大。研究显示,两者的CK升高程度不同,BMD的CK升高明显,可达正常参考值上限的25~200倍,LGMD2A仅1~80倍[8]。肌肉MRI中受累肌群有所不同,BMD腓肠肌内、外侧头均可受累,股四头肌股内侧肌和股外侧肌、 股中间肌受累明显,而LGMD2A腓肠肌外侧头和股四头肌的股外侧肌相对保留。BMD常累及心脏[9],心电图的经典表现包括:R波增高,下侧壁导联深 Q波和传导异常,超声心动图大多表现为左心室扩张[10]。而LGMD2A型较少累及心脏[11]。LGMD2A为常染色体隐性遗传,BMD为X连锁隐性遗传,家族史上表现亦不同。
本研究报告2例患者均为青年男性,隐匿起病,缓慢加重,四肢近端肌肉无力,以下肢无力为主,四肢腱反射不同程度减退或消失,中度增高,肌电图和肌肉活检均显示肌源性损伤,定位于肌肉,故临床初步诊断肌营养不良症。患者均有心脏受累,表现出心电图及心脏彩超异常,例2伴有双侧小腿假性肥大,易误诊为BMD。之后进行基于HTS的全外显子组测序,并采用Sanger测序验证测序找到的突变位点,qPCR对外显子水平的缺失进行验证,最终在CAPN3基因中找到了复合的杂合突变。该家系符合常染色体隐性遗传,结合临床表现和基因检查结果,两位患者最终确诊为LGMD2A。
本文报告了一对LGMD2A型兄弟,临床表现与BMD相似,容易误诊,提示临床表现、辅助检查及肌肉病理虽然对遗传性肌肉疾病的诊断有导向作用,但最终还需结合基因检测进行诊断。基因检测是诊断遗传性肌肉疾病的“金标准”,注意鉴别LGMD的分型[12]。本文报告了CAPN3基因新的复合杂合突变,增加了人群CAPN3基因突变的多样性和复杂性,证明了外显子测序分析对于小片段拷贝数变异分析的可行性和临床价值。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
临床输血与检验(2022年3期)2022-06-22
种子(2021年3期)2021-04-12
国际放射医学核医学杂志(2021年10期)2021-02-28
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
中国生育健康杂志(2018年6期)2018-11-13
食品安全导刊(2018年36期)2018-05-25
科技视界(2016年27期)2017-03-14
