
小麦TaPR1 基因启动子的克隆及启动活性分析
2022-02-21 09:34:16王丽珊王海燕刘大群
河北农业大学学报 2022年1期
王丽珊,王 珅,王 菲,王海燕,刘大群
(1.河北农业大学 植物保护学院/河北省农作物病虫害生物防治技术创新中心,河北 保定 071000; 2.河北省农林科学院 粮油作物研究所/河北省作物遗传育种实验室,河北 石家庄 050035)
PR1 是 病 程 相 关(Pathogenesis related,PR)蛋白家族成员,具有抗真菌、抗病毒功能,帮助植物抵抗逆境胁迫,同时也是病原物诱导的系统获得性 抗 性(Systemic acquire resistance,SAR)防 御信号获得的标志。刘玉晴等[1]在研究植物激素对水稻OsPR1A 表达的影响时发现,茉莉酸甲酯处理后的水稻幼苗在接种水稻白叶枯菌(Xanthomonas oryzae,Xoo)后6 d,OsPR1A 大量表达,病斑生长受到明显抑制;Van 等[2]发现拟南芥中编码PR1蛋白的22 个基因中,只有1 个PR1基因在受到细菌及激素诱导时提高表达;符晓等[3]发现大薯中6个DaPR1基因在炭疽菌侵染时表达量均显著提升。
本课题组前期利用Southern 杂交试验验证了TaPR1在小麦基因组中包含4 个拷贝,利用‘中国春’缺体- 四体系明确该基因定位于7D 染色 体[4];Gao[5]发现当受到信号分子脱落酸(Abscisic acid,ABA)诱导12 h 时,TaPR1基因出现明显的上调表达,表达量是0 h 的3.1 倍;水杨酸(Salicylic acid,SA)诱导后72 h,TaPR1基因出现表达高峰,是0 h 的5.4 倍。栗小英[6-7]发现TaPR1基因在叶锈菌侵染小麦12 h 后出现第1 个表达高峰,第2 个表达高峰出现在120 h,该基因在根、茎中的表达量远远低于在小麦叶片中的表达量,在小麦抗叶锈病近等基因系TcLr35 中,TaPR1的表达量随着植株生长而增加。张家瑞[8]利用基因枪将亚细胞定位重组体pCamA-TaLr35PR1-GFP 转化进入洋葱表皮细胞,发现TaPR1 蛋白定位在细胞外,且利用酵母系统验证TaPR1基因所携带的信号肽具有分泌活性。申松松等[9]利用GST-Pull down 及酵母双杂交技术筛选到谷氨酰氨合成酶(Glutamine synthetase)、细胞色素b6(Cytochrome b6)、应激蛋白(Stress protein)等小麦中与TaPR1 互作的蛋白。王菲等[10]利用VIGS 及体外抑菌试验证明TaPR1 能够有效抑制叶锈菌的生长,且TaPR1 能够通过与TaTLP1 的互作增强小麦抗叶锈病的能力。
前期已经明确TaPR1基因参与小麦抗叶锈病防御反应,然而对于调控TaPR1基因表达的转录调控因子了解甚少。因此,本研究拟通过克隆小麦TaPR1基因上游启动子序列,对其所包含的顺式作用元件进行分析预测,利用β-葡萄糖苷酸酶(β-glucuronidase,GUS)组 织 化 学 染 色、绿 色荧 光 蛋 白(Green fluorescent protein,GFP)验 证 2 200 bp、900 bp 及290 bp 3 种不同长度启动子序列启动活性,以期明确具有较强启动活性的区域用于后续试验,为进一步解析TaPR1基因转录调控机制奠定理论基础。
1 材料与方法
1.1 试验材料
植物材料为小麦抗叶锈病近等基因系材料TcLr19, 本 氏 烟; 植 物 表 达 载 体 为pBI121 和pCamA,均由河北农业大学植物病害生物防治和分子植物病理学实验室提供。无缝克隆试剂盒购自南京诺唯赞生物科技股份有限公司,GUS 染色试剂盒购自华越洋生物(北京)科技有限公司。
1.2 试验所需引物
本试验所有引物均由上海生工生物工程股份有限公司合成。

表1 用于启动子克隆与启动活性分析的特异性引物Table 1 Primers for promoter cloning and activation activity analysis
1.3 pTaPR1 的克隆及序列分析
本研究前期已经明确TaPR1基因定位于7D 染色体[4],根据‘中国春’小麦基因组数据库(http://plants.ensembl.org/Triticum_aestivum/Info/Index)信息,获得位于7D 染色体上TaPR1基因上游2 200 bp 序列,利用primer 5.0 设计特异性引物T-pro-TaPR1-F2200和T-pro-TaPR1-R。 以TcLr19 DNA 为模板进行扩增,连接载体pMD19-T 测序后序列比对,利用PlantCARE(http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)进行顺式元件的预测。根据PlantCARE 预测结果,结合前期研究[5]中TaPR1对ABA 及SA 的特异性响应,针对ABA 信号通路响应元件ABRE 以及SA 信号通路响应元件As-1,对pTaPR1进行5′端分段缺失。
1.4 植物表达载体的构建及烟草瞬时转化
参考无缝克隆试剂盒说明书完成启动活性验证 载 体pBI121-pTaPR12200、pBI121-pTaPR1900、pBI121-pTaPR1290、pCamA-pTaPR12200、pCamApTaPR1900及pCamA-pTaPR1290的构建。利用冻融法将连接产物转入农杆菌菌株GV3101,PCR 方法鉴定阳性克隆,挑取阳性克隆,利用含有利福平(50 μg/mL)及卡那霉素(50 μg/mL)的LBman 震荡培 养15 h,3 000 r/min 离 心15 min,重 悬 沉 淀 至OD600=0.8,注射本氏烟,避光培养48 h。
1.5 GUS 染色
设置未携带质粒的农杆菌GV3101为阴性对照,阳性对照携带pBI121 空载体。48 h 后取烟草叶片进行GUS 组织化学染色,利用打孔器对叶片取样,按照说明书配置GUS 染色工作液,将样品浸泡于GUS 染色液中,放置在37 ℃培养箱,染色期间全程避光。20 h 后利用75%酒精对样品进行脱色处理,至叶片叶绿素完全褪去,此时阴性对照呈白色,观察试验组叶片染色情况。
1.6 荧光观察
设置pCamA 空载体为阳性对照,未携带质粒的农杆菌GV3101 为阴性对照。48 h 后利用荧光显微镜观察烟草叶片中GFP 荧光表达情况。剪取适量被侵染烟草叶片制作玻片,利用尼康Ti2-U 倒置荧光显微镜对烟草叶片下表皮进行荧光观察,波长选择490 nm,曝光时长设置为600 ms,拍照记录细胞中绿色荧光表达情况。
2 结果与分析
2.1 pTaPR1 的克隆
利用引物T-pro-TaPR1-F2200和T-pro-TaPR1-R进 行 扩 增 后,获 得1 条 约2 300 bp 的 扩 增 条 带(见图1),与预期大小一致,成功构建重组载体T-pTaPR1,测序分析结果表明,序列中包括扩增用特异性引物T-pro-TaPR1-F2200和T-pro-TaPR1-R,全长共2 323 bp。BLAST 比对结果显示,扩增片段3′端113 bp 与TaPR1基因ORF 区5′端113 bp 相似性为100%,证明克隆得到的2 200 bp 位于TaPR1基因上游5′端,暂命名为pTaPR1,将此序列确定为后续研究目标。
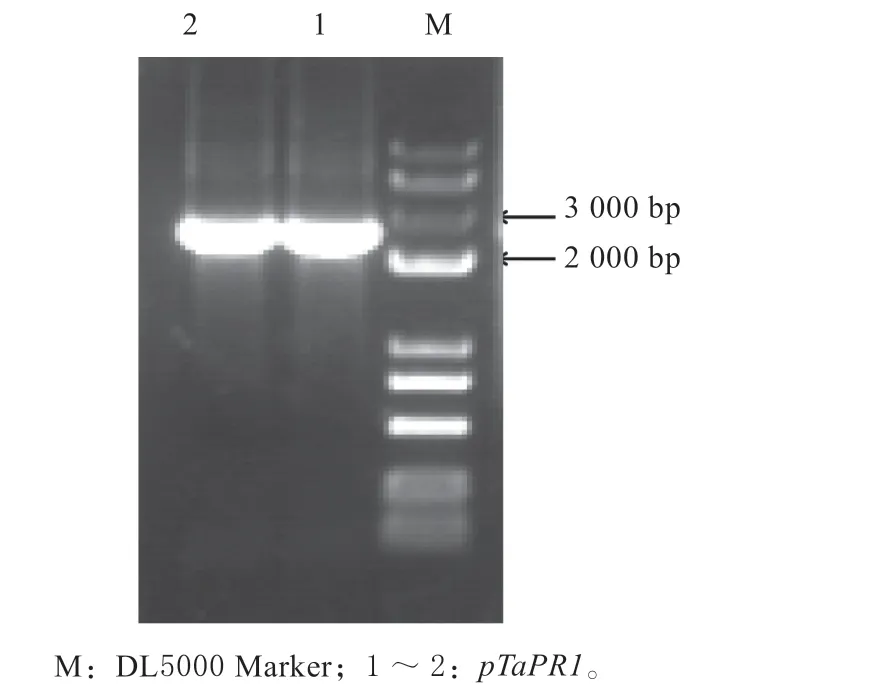
图1 pTaPR1 扩增结果Fig .1 Amplification results of pTaPR1
2.2 pTaPR1 顺式作用元件分析
利用PlantCARE 对序列进行顺式作用元件的分析,发现TaPR1基因上游启动子序列中包含启动子核心元件TATA-box、CAAT-box,还含有能够其他功能响应元件,如-1 960 bp、-1 482 bp、-436 bp、-284 bp 位置上响应SA 的AS-1 作用元件, -2 008 bp 位 置 上AACCTAA 序 列 的MYB 转 录 因子结合位点MRE,-2 200 bp 位置的WRKY 转录因 子 结 合 位 点W-box,-840 bp、-593 bp、-630 bp、-434 bp 位 置 的4 个ABA 响 应 元 件ABRE, -443 bp 的低温响应元件LTR,3 个无氧诱导响应元件ARE 分别在-1 816 bp、-1 347 bp、-867 bp,参与胚乳特异性表达的AACA_motif 作用元件在-220 bp,-164 bp 位置上游调控分生组织特异性表达元件CAT-box 等(见图2)。

图2 pTaPR1 顺式作用元件预测Fig . 2 Prediction of cis-acting elements in pTaPR1
2.3 pTaPR1 启动活性检测
2.3.1pTaPR1的分段 根据PlantCARE 预测结果,结合前期研究[5]中TaPR1对植物激素ABA 及SA的特异性响应,针对元件ABRE 以及AS-1 的位置对pTaPR1进行5′端缺失分段,将pTaPR1分为pTaPR12200、pTaPR1900及pTaPR12903 种不同区段。利用无缝克隆构建重组载体,电泳结果表明,各体系扩增产物大小分别为2 200、900 和290 bp(见图3),与预期大小一致。测序结果表明,成功构建重 组 载 体pBI121-pTaPR12200、pBI121-pTaPR1900、pBI121-pTaPR1290、pCamA-pTaPR12200、pCamApTaPR1900及pCamA-pTaPR1290,可用于后续试验。
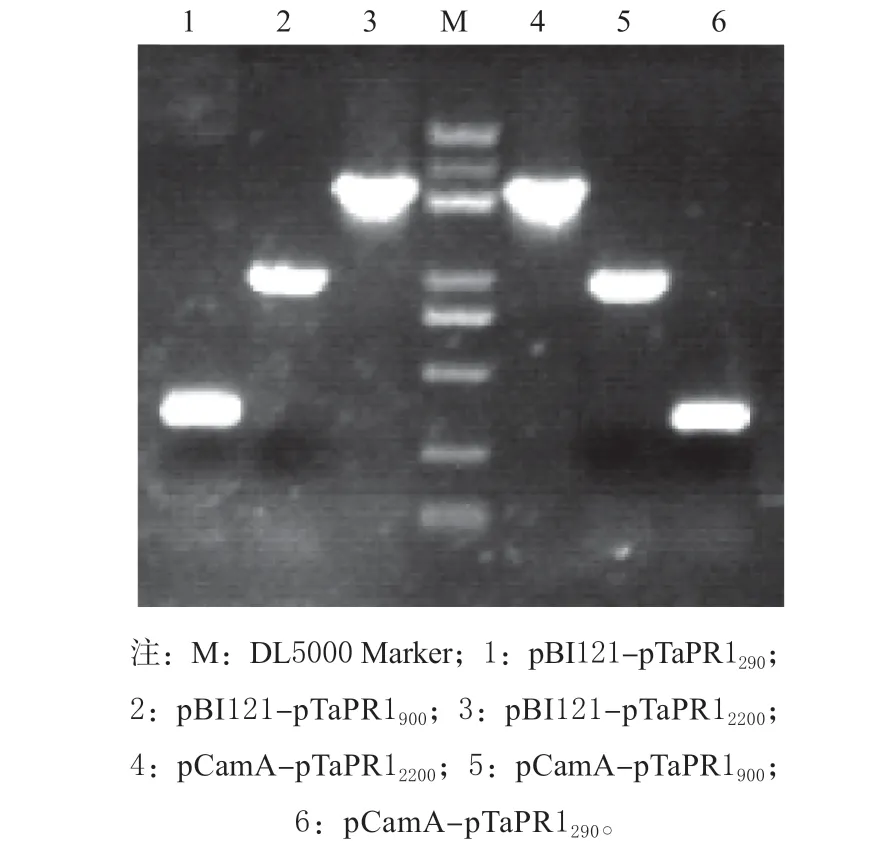
图3 重组载体菌液PCR 验证Fig. 3 PCR products of recombined vectors
2.3.2 GUS 活性检测 将重组载体pBI121-pTaPR12200、pBI121-pTaPR1900、pBI121-pTaPR1290分别转入农杆菌,注射烟草,GUS染色结果显示,阴性对照(未携带质粒的农杆菌GV3101)酒精脱色后叶片呈白色,表明烟草叶片中无GUS基因本底表达;携带质粒pBI121 的农杆菌为阳性对照,该组烟草叶片呈蓝色,表明GUS基因在35S 启动子的启动下,在烟草叶片中正常表达;携带重组载体pBI121-pTaPR12200和pBI121-pTaPR1900的农杆菌侵染烟草叶片,脱色后的叶片显蓝色,证明2 200 bp 和900 bp 的pTaPR1具有启动活性(见图4);携带重组载体pBI121-pTaPR1290的农杆菌侵染烟草叶片,脱色后叶片显白色,证明290 bppTaPR1无法启动下游GUS基因表达。

图4 pTaPR1 驱动GUS 基因的表达验证Fig. 4 Validation of the expression of GUS gene driven by pTaPR1
2.3.3 GFP 荧光观察 将重组载体pCamA-pTaPR12200、pCamA-pTaPR1900、pCamA-pTaPR1290分别转入农杆菌,注射烟草,GFP 荧光观察结果显示,阴性对照(未携带质粒的农杆菌GV3101)叶片在荧光场下无可见绿叶荧光,表明烟草叶片中GFP基因无本底表达;携带质粒pCamA 农杆菌为阳性对照,叶片在荧光场可观察到绿色荧光,细胞核及细胞轮廓清晰可见,表明GFP基因在35S 启动子的启动下,在烟草叶片中正常表达;当农杆菌携带重组载体pCamApTaPR12200和pCamA-pTaPR1900侵染烟草叶片,荧光场中可观察到绿色荧光,GFP基因正常表达,pTaPR12200和pTaPR1900具有启动活性;当农杆菌携带重组载体pCamA-pTaPR1290侵染烟草叶片,无法观察到绿色荧光,290 bppTaPR1不能激活GFP基因表达,pTaPR1290无启动活性(见图5)。
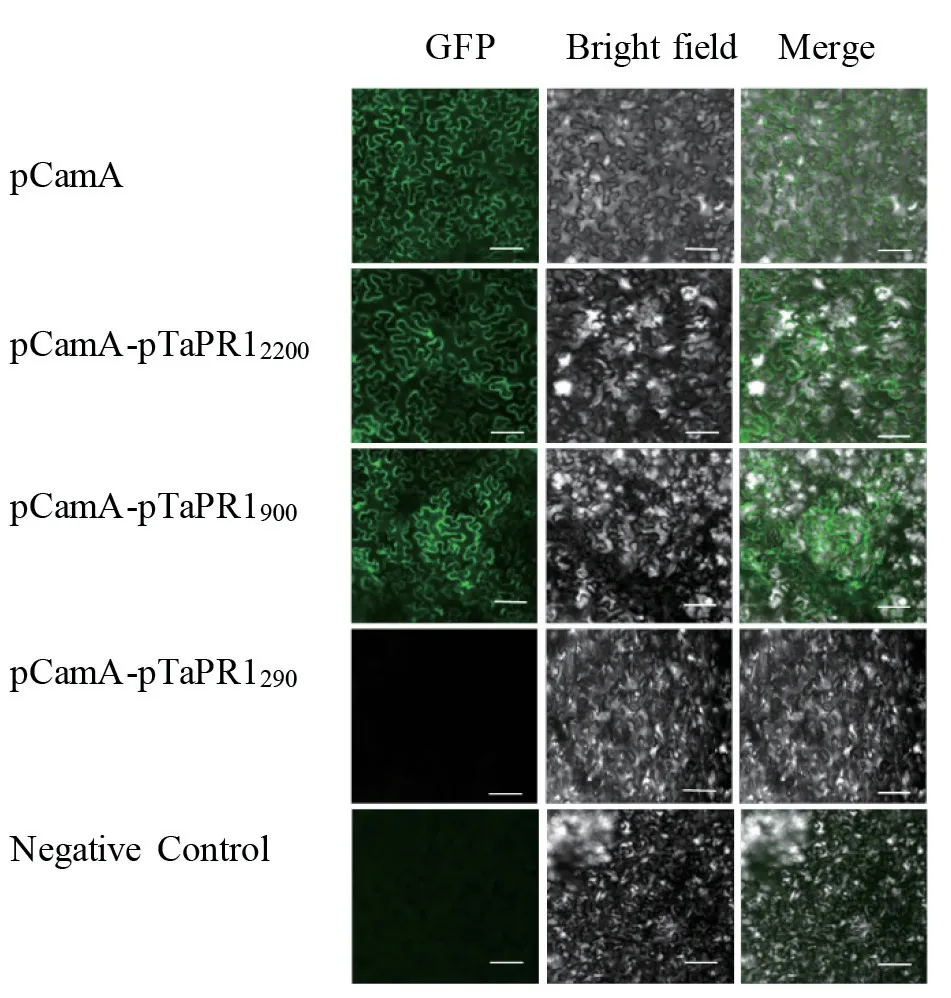
图5 烟草叶片的GFP 荧光观察Fig. 5 GFP fluorescence observation of tobacco
3 讨论与结论
5′端缺失分段是寻找启动子启动活性关键区段的有效方法。董向向等[11]克隆获得草莓开花控制基因ARF4基因启动子,在顺式作用元件预测的基础上进行缺失分段,利用GUS染色分析启动子表达的特异性,确定生长素和赤霉素是影响ARF4基因启动子转录活性的因素;石翠翠等[12]利用启动子分段克隆的方法,寻找拟南芥AtRPK1启动子启动关键区域,利用5′端缺失的方法得到不同长度的片段,分别构建表达载体,转入拟南芥验证启动活性,发现809 bp 启动子启动的基因在各组织中均有表达,具有较高启动表达的能力;柴春月等[13]研究发现GmDRRP基因表达受激素的抑制,克隆上游1 500 bp 启动子区,发现该区域包含多个已知逆境响应相关的顺式元件,启动活性验证试验表明拟南芥接种疫霉菌0.5 hGmDRRP启动子快速相应诱导,2 h 后报告基因表达量显著上升,222 bp 片段对疫霉的诱导响应能力是全长启动子的34%;杨少华等[14]研究发现甘薯IbMYB1基因启动子的关键区域位于 -2 183 ~-1 000 bp 区 段; 李 静[15]发 现 大 豆GmCBL6基因启动子关键区域位于-969 ~-769 bp区段。本试验为验证TaPR1基因启动子具有启动活性的具体功能区域,利用GUS 染色及GFP 荧光观察 对pTaPR12200、pTaPR1900和pTaPR1290进 行 启 动活性验证,结果发现仅有pTaPR12200和pTaPR1900能够驱动下游报告基因的表达,表明TaPR1基因启动子活性的主要功能区域位于-2 200 ~-290 bp 区段。
启动子在上游转录调控区中含有特殊的顺式调控元件,这些调控元件通常可通过与某些特定的刺激信号分子结合,从而赋予启动子特定的调控和表达方式。顺式作用元件As-1 在基因响应SA 诱导时发挥重要作用,1989 年,Eric[16]发现As-1 元件是决定CaMV35S 启动子启动活性的关键区域;Qin 等[17]利用启动子区域点突变构建GUS基因融合表达载体,通过验证不同激素诱导情况下转基因植株中GUS基因的表达,发现As-1 顺式作用元件是35S 启动子响应SA 诱导的关键元件。在目前确认到的具有启动活性的pTaPR1 中,存在3 个SA 信号通路TGA 转录因子结合位点As-1,猜测是该顺式作用元件在调控TaPR1基因转录过程中发挥重要作用,本研究结果将为后续解析TaPR1 基因转录调控机制奠定基础。
猜你喜欢
奥秘(创新大赛)(2023年3期)2023-05-06 01:48:20
环球时报(2022-09-20)2022-09-20 15:18:57
今日农业(2020年24期)2020-12-15 16:16:00
浙江中西医结合杂志(2017年2期)2017-01-12 18:23:59
现代工业经济和信息化(2016年2期)2016-05-17 05:34:16
兽医导刊(2016年12期)2016-05-17 03:51:50
当代化工研究(2016年9期)2016-03-20 16:22:08
电子工业专用设备(2015年4期)2015-05-26 09:10:40
汽车维修与保养(2015年8期)2015-04-17 03:33:01
现代检验医学杂志(2015年4期)2015-02-06 02:02:06
