
GCH1基因突变致多巴反应性肌张力不全一例
2022-02-16 23:20黄健星林小群黄洁馨
新医学 2022年1期
黄健星 林小群 黄洁馨
【摘要】 多巴反应性肌张力不全(DRD)是一种常染色体显性或隐性遗传运动障碍疾病,病理特征为黑质纹状体多巴胺含量减少,主要表现为儿童期肌张力障碍, 小剂量左旋多巴可以改善症状。三磷酸鸟苷环化水解酶1(GCH1)基因是其致病基因之一。该文报道1例GCH1基因突变所致的DRD患儿,其以步态异常为主诉,行走时出现肌张力不全姿势,症状晨轻暮重,经基因检测证实其GCH1基因出现杂合性突变。接受小剂量左旋多巴治疗后患儿神经系统体征基本消失,于小儿神经科门诊随诊2年,运动功能良好。临床上对于病因不明的肌张力障碍患儿, 需警惕DRD的可能, 尽早识别和治疗可明显改善预后,预防残疾的发生。
【关键词】 多巴反应性肌张力不全;三磷酸鸟苷环化水解酶1基因;左旋多巴
Dopa-responsive dystonia induced by GCH1 gene mutation: a case report Wong Kin-Sing, Lam Sio-Kuan, Wong Kit-Hing.department of pediatrics, Kiang Wu Hospital, Macao Special Administrative Region, China
Corresponding author,Wong Kin-Sing,E-mail: star-wong311@hotmail.com
【Abstract】 Dopa-responsive dystonia(DRD) is an autosomal dominant or recessive dyskinesia, which is pathologically characterized with reduced amount of dopamine in the substantia nigra and striatum. DRD is mainly manifested with dystonian in early childhood. Low-dose levodopa can mitigate relevant symptoms. Guanosine triphosphate cyclohydrolase I (GCH1) gene is the pathogenic gene of DRD. In this article, 1 child diagnosed with DRD induced by GCH1 gene mutation was reported. Gait abnormality was the chief complaint. The child showed dystonic posture when walking. These symptoms were mitigated in the morning and aggravated during night. Gene detection confirmed the incidence of heterozygous mutation of GCH1 gene. The neurological symptoms of this child were basically cured after low-dose levodopa therapy. During 2-year follow-up at Department of Pediatric Neurology, motor function was restored to normal. In clinical practice, the possibility of DRD should be considered for pediatric dystonia with unknown causes. Prompt diagnosis and treatment can significantly improve clinical prognosis and prevent the incidence of disability.
【Key words】 Dopa-responsive dystonia; GCH1 gene; Levodopa
多巴反應性肌张力不全(DRD, 又称Segawa病),属于神经传导物质病变, 是累及神经系统的罕见疾病。儿童时期往往以步态异常为首发症状,给予小剂量左旋多巴症状可好转。遗传方式多为常染色体显性遗传,三磷酸鸟苷环化水解酶1(GCH1)基因是DRD的致病基因(GCH1基因突变也可以表现为常染色体隐性遗传),其他基因例如酪氨酸羟化酶 (TH)、 墨蝶呤还原酶(SPR)突变则表现为常染色体隐性遗传。本文报道1例DRD,基因测序显示其存在GCH1基因突变,具体如下。
病例资料
一、一般情况
患儿女,8岁4个月,因步态异常8月余于2019年7月30日入住澳门镜湖医院儿科。患儿缓慢起病,无明显诱因下于行走时出现肌张力不全姿势,表现为右下肢屈曲内翻,伴右上肢僵硬,晨轻暮重,经晚上休息后早上好转,无伴头痛、抽搐。患儿出生史正常,既往史无特殊, 自幼运动、语言发育正常,智力正常,学习成绩可。其父亲有轻微步态异常10余年,未予重视,此前未曾就诊,其母亲身体健康。
二、体格检查、实验室及辅助检查
体格检查:发育正常,无特殊面容及皮肤胎记,心、肺、腹无特殊,颅神经检查无特殊,四肢各关节未见畸形,脊柱未见异常,肌容积正常,四肢肌力Ⅴ级,四肢肌张力正常,生理反射正常,右侧髁阵挛阳性,病理反射未引出,脑膜刺激征阴性,共济试验完成准确。实验室及辅助检查:外周血常规正常,ESR不高,肝肾功能、心肌酶、电解质及血气分析正常,催乳素正常,铜蓝蛋白正常,血氨、乳酸不高,RF、抗核抗体、抗双链DNA及抗O均阴性,血串联质谱氨基酸及肉碱谱正常,尿气相色谱有机酸正常。双下肢立位X线片检查未见异常。颅脑及脊髓MRI平扫及增强未见异常。上下肢神经运动感觉传导速度检测正常。
三、诊治经过
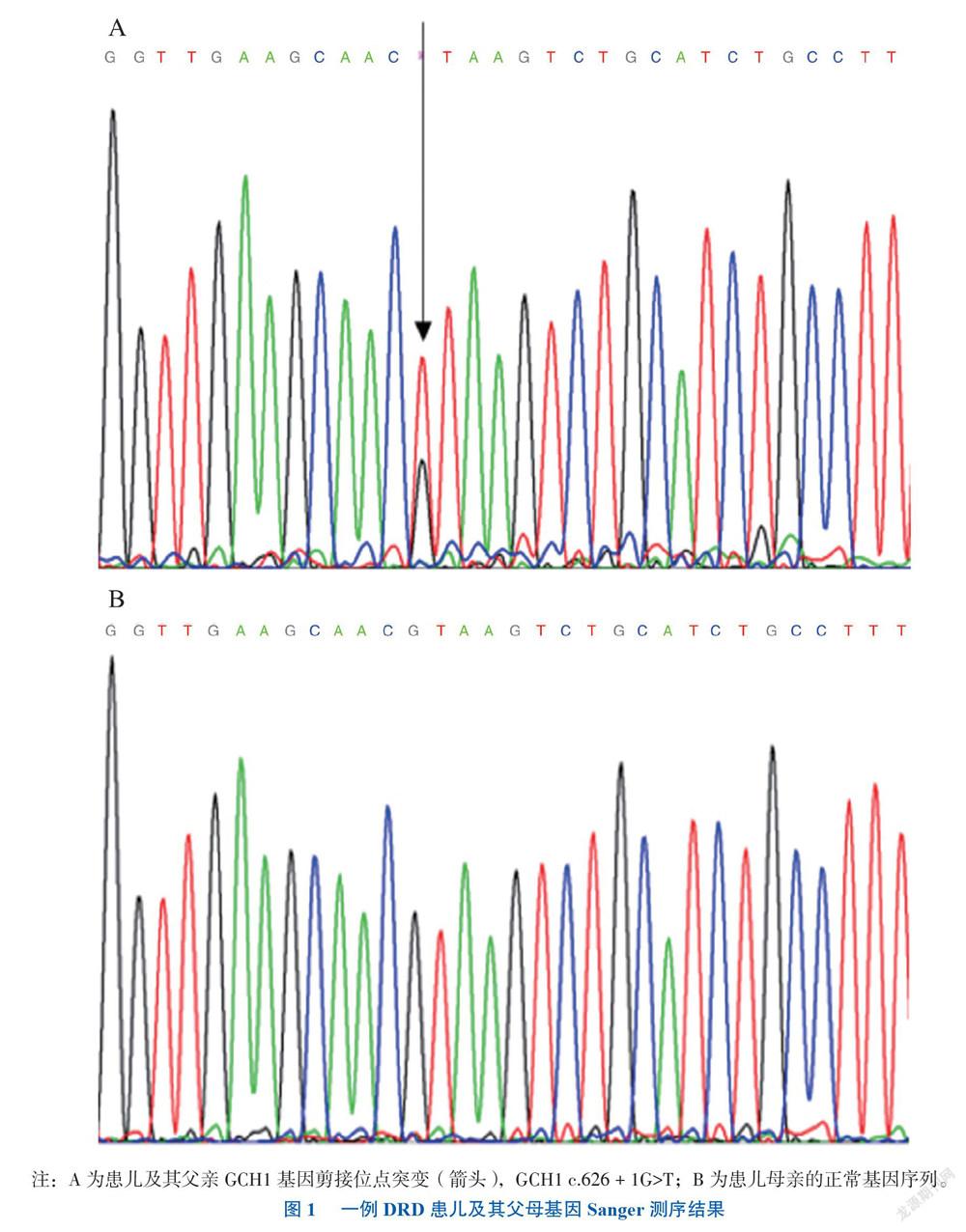
根据患儿临床表现高度怀疑DRD,予其小剂量左旋多巴作诊断性治疗,剂量为40 mg/d[相当于1.5 mg/(kg·d)],治疗1周后患儿症状好转,2019年8月基因外显子测序+Sanger测序提示GCH1基因杂合性剪接位点突变c.626 + 1G>T,其父亲有同样的突变而母亲没有,见图1。患儿诊断DRD明确,嘱其长期服用左旋多巴及在小儿神经科门诊接受长期随诊,随诊2年余时其神经系统体征基本消失,运动功能良好,病情稳定。
讨 论
DRD于1976年由Segawa首次描述,故又称为Segawa病[1]。其临床表现多样,主要表现为肌张力不全及帕金森样症状,以晨轻暮重现象、小剂量多巴治疗有显著且持续的疗效为特点[2]。文献报导部分病例可出现不典型症状,例如新生儿期起病、严重运动障碍(吸吮、吞咽困难,严重肌张力低下,眼睑下垂,抽搐,智能倒退,疲乏,激惹,反复体温过低等),被称为DRD-plus[3]。本例DRD的诊断依据如下:①学龄期女性,缓慢起病;②行走时可见肌张力不全姿势,晨轻暮重;③右侧髁阵挛阳性;④其父亲有轻微步态异常;⑤小剂量左旋多巴治疗有明显效果;⑥患儿及其父亲基因检测结果均为GCH1 基因杂合性剪接位点突变 c.626 + 1G > T。
DRD的遗传方式多为常染色体显性遗传或隐性遗传, 隐性遗传患者症状较显性者严重。除了GCH1之外,其他基因如TH、SPR突变也可致DRD或DRD-plus[4-5]。相同的基因突变可有临床表型异质性,症状的严重程度与突变的数量、酶活性减少的程度有关。本例患儿GCH1基因突变 c.626 + 1G > T,为杂合性突变, 该基因为剪接位点(+1)突变, 发生功能缺失, 是导致DRD的致病因素, 正常人携带频率为0, 有权威的研究和数据库支持该突变是致病的, 且多项计算机模拟计算预测此剪接位点突变为有害的影响, 根据美国医学遗传学与基因组学学会指南,判定该突变位点为致病性突变(PSV1+PM2+PP5+PP3)。GCH1基因突变典型表现为年轻患者、以下肢肌张力不全为首发症状、无明显认知功能受累等,本例患儿临床表现和文献报导相符[6]。
与帕金森病黑质纹状体多巴胺神经元进行性变性死亡不同,DRD没有细胞减少[7]。DRD属于神经传导物质病变,是由于脑内神经传导物质的生化改变所致。GCH1参与合成四氢生物喋呤(BH4)的首个步骤,BH4是TH的辅酶,TH把酪氨酸合成多巴胺,因此,GCH1缺乏会阻碍TH将酪氨酸转化为多巴胺[6]。BH4也是苯丙氨酸羟化酶的辅酶,如肝脏缺乏此酶,会导致苯丙酮尿症(PKU) [8]。但GCH1缺乏不会导致PKU,原因是不同器官表达不同比例的正常/突变mRNA,这使不同器官的GCH1活性有所不同,DRD患者的GCH1缺乏选择性地影响脑部而不影響肝脏。故DRD患者没有PKU的症状。
儿童肌张力不全,临床上需要排除获得性因素,包括围产期缺血缺氧、颅脑外伤、颅内感染、脑血管意外、药物中毒及脑肿瘤[9]。病因不明者可诊断性试用左旋多巴以排除DRD[10]。虽然DRD不常见,但DRD患儿经小剂量左旋多巴治疗往往获得好转,因此对于表现为特发性肌张力不全者,在排除上述获得性病因后,非常值得试用左旋多巴以进一步明确是否为DRD。DRD患儿需要长期使用左旋多巴,开始剂量通常为1 mg/(kg·d),逐渐缓慢上调剂量以获得最佳效果, 目标剂量可达5~10 mg/(kg·d), 部分患儿无需达到此剂量即可获得显著效果[11]。
DRD容易被误诊漏诊,部分患者起病至获得诊断的时间可长达十余年[12]。如患者未能获得适当治疗,基底节长期缺乏多巴胺会致调节运动功能出现混乱,DRD的肌张力不全及帕金森样症状渐渐加重,对左旋多巴的治疗最终不再有反应。故临床医师应提高对DRD的认识水平,及早实施诊断与治疗,这将对此病患儿的预后有很大帮助,可有效预防残疾的发生。
参 考 文 献
[1] Segawa M, Hosaka A, Miyagawa F, et al. Hereditary progressive dystonia with marked diurnal fluctuation. Adv Neurol,1976,14:215-233.
[2] Furukawa Y. GTP Cyclohydrolase 1-Deficient Dopa-Responsive Dystonia. 2002 Feb 21 [updated 2019 Jan 24]. In: Adam M P, Ardinger H H, Pagon R A, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2021. PMID: 20301681.
[3] Cherian A, Paramasivan N K, Divya K P. Dopa-responsive dystonia, DRD-plus and DRD look-alike: a pragmatic review. Acta Neurol Belg,2021,121(3):613-623.
[4] Dong H Y, Feng J Y, Yue X J, et al. Dopa-responsive dystonia caused by tyrosine hydroxylase deficiency: three cases report and literature review. Medicine (Baltimore),2020,99(33):e21753.
[5] Froukh T. Genetic study in a family with dopa-responsive dystonia revealed a novel mutation in sepiapterin reductase gene. Pak J Med Sci,2019,35(6):1736-1739.
[6] Yoshino H, Nishioka K, Li Y, et al. GCH1 mutations in dopa-responsive dystonia and Parkinson’s disease. J Neurol,2018,265(8):1860-1870.
[7] 张旺明,徐如祥.帕金森病的病理生理研究进展.新医学,2002,33(5):165-167.
[8] van Wegberg A M J, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis,2017,12(1):162.
[9] Albanese A, Di Giovanni M, Lalli S. Dystonia: diagnosis and management. Eur J Neurol,2019,26(1):5-17.
[10] Randby H, Salvador C L, Oppebøen M, et al. Dopa-responsive dystonia. Tidsskr Nor Laegeforen,2018,138(19).
[11] Luc Q N, Querubin J. Clinical management of dystonia in childhood. Paediatr Drugs,2017,19(5):447-461.
[12] Kim W, Cho J S, Shim Y K, et al. Early-onset autosomal dominant GTP-cyclohydrolase I deficiency: diagnostic delay and residual motor signs. Brain Dev,2021,43(7):759-767.
(收稿日期:2021-07-25)
(本文編辑:洪悦民)
猜你喜欢
中国医学创新(2022年17期)2022-07-10
文萃报·周二版(2021年47期)2021-12-14
保健与生活(2021年15期)2021-08-16
电脑报(2020年40期)2020-11-06
三农资讯半月报(2020年11期)2020-06-21
家禽科学(2019年4期)2019-07-08
电脑知识与技术(2018年19期)2018-11-01
家庭百事通·健康一点通(2018年9期)2018-10-12
家庭百事通·健康一点通(2017年8期)2017-08-18
中外医学研究(2016年35期)2017-02-28
