
高效液相色谱柱后衍生荧光法测定柑橘中8种氨基甲酸酯类农药残留
2022-02-06 07:51:26杜鑫
农药科学与管理 2022年11期
杜 鑫
(南充农产品质量监测检验中心,四川 南充 637000)
氨基甲酸酯类农药是以甲酸酯为前体化合物发展而来的一类高效、广谱杀虫剂,通过抑制昆虫体内乙酰胆碱酶和羧酸酯酶活性,使得昆虫神经功能紊乱而死,具有残留期短、易代谢、低毒效和择性强等特点,主要应用于果树、蔬菜及粮食等农作物的虫害防治[1-2]。目前,这类农药的仪器检测方法主要有气相色谱法[3-4]、气相色谱-串联质谱法[5-6]、液相色谱-紫外光度法[7]、液相色谱-柱后衍生荧光法[8-10]、液相色谱-串联质谱法[11-13]。由于氨基甲酸酯类农药具有热不稳定性,气相色谱法受到一定限制。液相色谱-紫外光度法检测氨基甲酸酯类农药偏少,选择性较差,灵敏度低、较易出现假阳性。质谱联用技术广泛应用于多组分农药的高通量、高灵敏度定性及定量分析,已成为农药多残留检测的主导技术和未来发展的趋势[14-16],但因其价格昂贵、操作复杂、尚不便于基层检测实验室应用普及。本研究基于液相色谱-柱后衍生荧光检测系统,建立了检测柑橘中8种氨基甲酸酯类农药残留的分析方法,优化了系统检测条件,极大地提高了分析方法的效率、选择性和灵敏度,以期为基层检测机构提供技术参考和借鉴。
1 材料和方法
1.1 试剂耗材 涕灭威亚砜、涕灭威砜、涕灭威、灭多威、三羟基克百威、克百威、甲萘威、异丙威均购于农业农村部环境质量监督检验测试中心(天津),浓度均为1 000mg/L,用纯甲醇稀释配制成浓度均为100.0mg/L标准储备液,避光保存于4℃冰箱内。柱后衍生试剂为水分解碱液为0.05mol/L NaOH溶液(cat.No CB130),OPA稀释剂(cat.No CB910),邻苯二甲醛(O-Phthaladehyde,OPA,cat.NO 0120),巯基乙醇(Thiofluor,cat. NO 3700-2000),均购于美国Pickering公司。
固相萃取小柱(Agilent,SPE-NH2小柱 500mg/6mL,货号5982-1865),“CNW”陶瓷均质子(货号CA8A50)和固相萃取装置(Agilent);甲醇,乙腈,二氯甲烷等均为色谱级;NaCl为分析纯试剂。50mL离心管,PTFE 0.22μm针筒式微孔滤膜。
1.2 仪器设备 Waters Acquity UPLC H-Class超高效液相色谱仪(配有Waters Reagent Manager柱后衍生反应装置),2475型荧光检测器,ML802型分析天平,GD16plus高速研磨均质仪,3-18KS型高速冷冻离心机,RE-2000A型旋转蒸发仪,VORTEX 3型旋涡混合器,HR386破壁机。
1.3 样品制备 取有代表性的柑橘(爱媛、春见、柠檬等)全果样品带皮切碎,采用四分法缩分后约1kg,放入破壁机中,高转速(30 000 r /min)匀浆均质后装入洁净的聚乙烯塑料样品盒中,密封后于-18 ℃冰箱保存(使用前先解冻至室温)。
1.4 样品提取 称取10g(精确至0.01g)柑橘样品于50mL塑料离心管底部,依次加入1颗陶瓷均质子,10.00mL乙腈,5 ~ 7g 氯化钠,拧紧离心管盖,放入低温(5℃)高速研磨均质仪中3 000r/min均质提取3min,然后5 000r/min离心3min。吸取上清液5.00mL至250mL锥形瓶中,40℃水浴减压旋转蒸发浓缩至近干,加入4mL甲醇+二氯甲烷(5:95,V:V)溶解残余物,涡旋混匀后待净化。
1.5 样品净化 将固相萃取氨基小柱(SPE-NH2)用4mL甲醇-二氯甲烷溶液(5:95,V:V)预淋洗,当液面到达柱筛板顶部时,立即加入上述待净化溶液,用50mL茄形瓶收集洗脱液,用3mL甲醇+二氯甲烷(5:95,V:V)涮洗锥形瓶后过柱,并重复1次,收集的全部洗脱液于30℃水浴旋转蒸发浓缩至近干,准确加入2.50mL甲醇-水(4:6,V:V),涡旋混匀,用0.22μm针孔式微孔滤膜过滤,待测。
1.6 色谱条件 采用Waters XBridge C18色谱柱(4.6mm×150mm,4.5μm),柱温为40℃,样品室温度为20℃,进样体积为20.00μL。流动相为甲醇(A)、纯水(B)和乙腈(C)的混合溶液,按表1中的流动相梯度洗脱条件对8种氨基甲酸酯类农药进行洗脱分离。实样测定过程中,如遇样品中的杂质峰对目标物色谱峰有干扰,适当调整流动相的梯度洗脱比例,使两者达到基线分离。
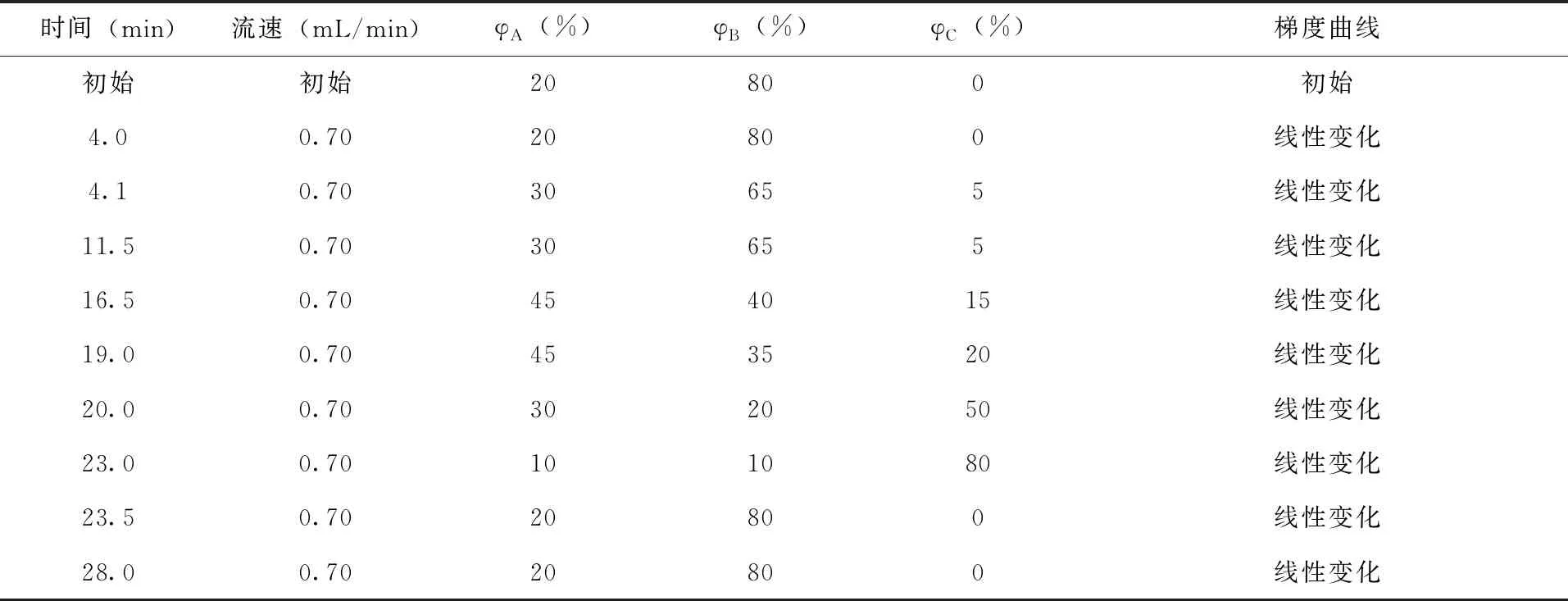
表1 高效液相色谱流动相组成及梯度洗脱条件
1.7 柱后衍生荧光检测系统 建立检测8种氨基甲酸酯类农药的高效液相色谱-柱后衍生荧光检测系统(图1),每一种氨基甲酸酯农药被色谱柱梯度洗脱分离后,在较高温度下与强碱NaOH溶液在水解反应盘管中发生水解反应生成甲胺,甲胺与邻苯二甲醛和巯基乙醇的混合溶液(OPA)在衍生反应盘管中反应转变成具有强荧光相应的异吲哚衍生物,进入荧光检测器,在激发波长330nm,发射波长465nm条件下进行检测。水解试剂和衍生试剂管线的流量均为0.20mL/min,水解反应盘管的加热器温度为80℃,衍生反应盘管温度为室温。
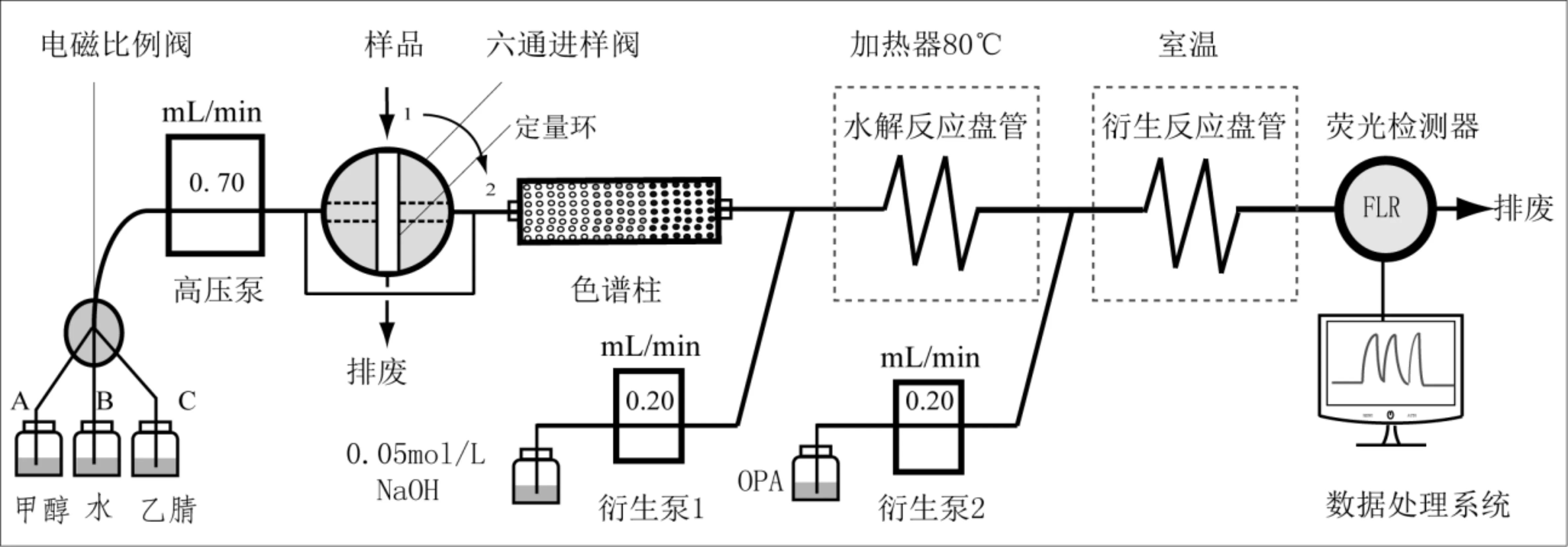
图1 高效液相色谱-柱后衍生荧光检测系统
1.8 衍生试剂配制 水解试剂用0.05moL/L NaOH溶液(CB130),使用前将衍生泵1的吸液管滤头插入后即可使用。衍生反应试剂为OPA溶液(现配现用),称取0.10g 邻苯二甲醛固体,加入10mL纯甲醇溶解,1.0g巯基乙醇固体用适量OPA稀释剂(CB910)溶解,将完全溶解后的邻苯二甲醛和巯基乙醇溶液倒入OPA稀释剂瓶中,盖上瓶盖,轻微振荡混合均匀,再插入衍生泵2的吸液管。
2 结果讨论
2.1 分离条件优化 试验初步选取甲醇(A)、纯水(B)和乙腈(C)为流动相,试样定容剂为甲醇-水(5:5,V:V)混合溶液,进样体积为20.00μL,衍生泵流量均为0.30mL/min。由于涕灭威亚砜和的涕灭威砜具有非常强的极性,化学性质非常相似,洗脱分离过程中两者不容易达到基线分离,且易与样品中含有的强极性干扰物的色谱峰重叠,故优先选用了极性较强但洗脱能力较弱的甲醇+水(2:8,V:V)作为流动相的起始梯度。当样品中的强极性干扰物与目标物基线分离后,增加流动相中乙腈比例,通过观察8种农药的保留时间、峰形、相邻峰之间的分离度、荧光响应信号强度等来优化液相色谱流动相的洗脱程序,结果(表1)。该条件下能够对8种氨基甲酸酯农药达到基线分离,图2为混合标准溶液的标准曲线色谱图,22min内所有目标物全部洗脱分离出来。可见,用甲醇-乙腈-水混合体系进行梯度洗脱时,提高了流动相洗脱强度和效率,有效改善了色谱峰峰形,增加了色谱峰响应信号,提高了色谱峰分离度,极大地缩短了样品的上机检测时间。
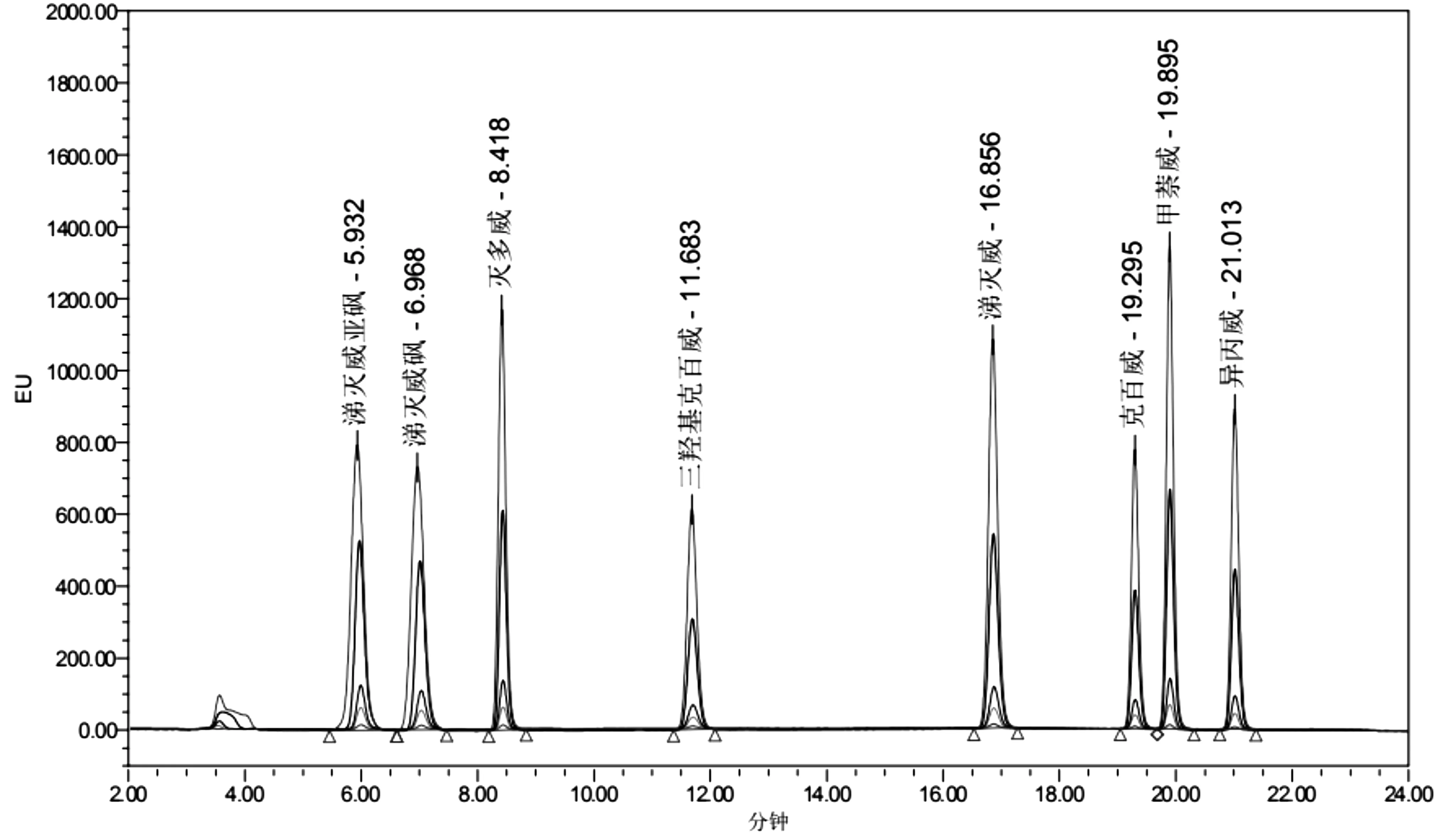
图2 8种氨基甲酸酯类农药的混合标准溶液色谱图
2.2 样品提取剂优化 氨基甲酸酯类农药难溶于水,较易溶于甲醇、乙腈、丙酮、乙酸乙酯等有机溶剂。因柑橘样品中含有糖类、有机酸、维生素、色素及大量水分,为更充分地提取待测药物,试验选用能与水互溶的乙腈和甲醇为提取剂。试验发现:甲醇提取样品后,有机相和水相难于彻底分离;乙腈对不同极性农药均有很好的提取效果,利用盐析法又能与水两相分离,还可以除掉样品提取液中的有机酸物、糖类及维生素等大部分杂质,有利于下一步净化。经优化后,选用10.00mL乙腈为提取剂。
2.3 提取方式优化 初步选用超声振荡提取、均质机匀浆提取、均质仪振荡提取方式对样品进行处理。试验发现:用高频超声波振荡提取样品超过40min后,水温可升至60℃左右,可能会导致部分目标物农药损失,每小时仅能处理样品约40个;每个样品如用高速匀浆机匀浆提取后,需要用水、丙酮+水(1:1,V:V)、水依次交替清洗匀浆机刀头,溶剂耗损量大,过程耗时长,不便于快速提取大批量样品,也容易造成样品间的交叉污染;采用高速研磨均质仪提取后,避免了样品间的交叉污染,每小时可处理约200个样品,极大地提高了样品提取效率。综合考虑,选用高速振荡均质仪振荡提取样品。
2.4 洗脱剂优化 据文献研究[9-10],SPE-NH2小柱可有效地除掉提取液中的碳水化合物、有机酸、糖分、酚类、色素及极性化合物等干扰杂质。用相同空白基质配制目标物混合标准溶液,用SPE-NH2对其进行净化,以用不同体积比(1:99、3:97、5:95、7:93、10:90)的甲醇+二氯甲烷混合溶液为洗脱剂,考察了其对目标物洗脱效果的影响。结果表明:当洗脱剂中甲醇体积分数增加,回收率基本稳定。当用6mL甲醇-二氯甲烷(5:95,V:V)为洗脱剂,分2次洗脱,目标药物附近的干扰峰较少且回收率接近100%。
2.5 样品溶剂优化 样品溶剂不仅要完全溶解样品中的待测农药,也要有利于减少溶解样品中的干扰物,便于上机分析检测。以纯甲醇为样品溶剂时,所得的色谱图(图3)。可以发现,较早洗脱出的涕灭威亚砜和涕灭威砜的色谱峰扭曲和变形了,而其余较之晚出峰的其他6种农药组分的色谱峰正常。涕灭威亚砜和涕灭威砜浓度越低,色谱峰展宽、扭曲、变形和分叉越严重,这是因为用强溶剂纯甲醇溶解样品时,样品溶剂在柱内可以看作流动相的一部分,溶解于强溶剂的洗脱带,一部分样品会被强溶剂迅速洗脱出色谱柱,另一部分在洗脱过程中溶于流动相,被流动相洗脱出,从而造成了色谱峰的展宽、扭曲和分叉,导致分析方法的准确度和精密度下降。这种现象符合溶剂效应[17],当样品溶液的溶剂强度强于流动相强度时,造成较早洗脱的峰前沿、扭曲或开叉,而较晚洗脱的色谱峰则正常。
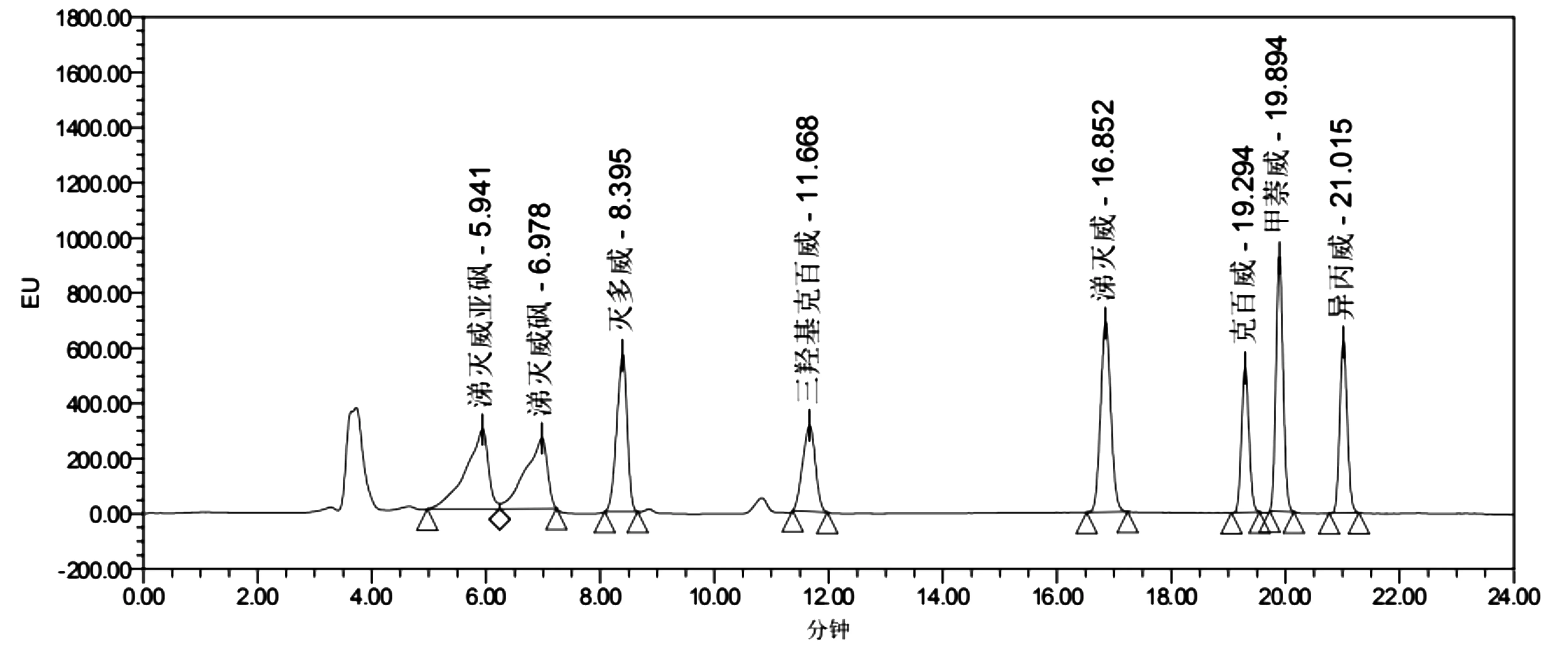
图3 样品溶剂对8种氨基甲酸酯农药色谱峰的影响(0.050mg/L)
另外,采用减小试样进样体积或用流动相梯度洗脱的初始比例溶液来溶解样品可以减小或者消除“溶剂效应”。当进样体积减小后,目标物的荧光响应信号值减小,方法灵敏度降低;用流动相的初始比例甲醇+水(2:8,V:V)溶解样品时,由于氨基甲酸酯类农药易溶于有机溶剂,不易溶于水,可能导致样品溶剂不能完全溶解样品中的强保留目标物,导致重现性变差及回收率降低。经优化,选用甲醇+水(4:6,V:V)为样品溶剂,不仅有效改善了加标样品中涕灭威亚砜和涕灭威砜的峰形,还降低了基线噪音,提高了分离度和荧光响应值。
2.6 水解反应温度优化 考察了柱后衍生系统中水解反应盘管的加热器(图1)在30℃~100℃温度范围内对目标物荧光响应值的影响。结果表明,当加热器温度>70℃时,8种农药的荧光响应值均达到最大值并趋于稳定,表明色谱梯度洗脱分离后的目标物与水解试剂NaOH完全反应,最终确定水解反应加热器温度为80℃。
2.7 衍生试剂流量及稳定性 当衍生试剂浓度一定时,其流量不仅决定从色谱柱中洗脱出的目标物与衍生试剂的反应程度,还会影响目标物的峰展宽、峰高、分离度及分析速度。在0.10~0.50mL/min范围内,考察了衍生试剂流量的影响,结果表明:当流量为0.20~0.40mL/min时,8种氨基甲酸酯农药与衍生试剂均完全反应,产物的荧光响应信号基本不变,峰形和分离度很好,为节省衍生试剂,确定衍生试剂流量均为0.20mL/min。另外,当衍生试剂OPA使用时间超过5d,溶液略微发黄,目标物的荧光响应值也会降低,故OPA衍生试剂一定要现配现用,且避免阳光直射,建议放入瓦楞纸箱中,并在旁边放入冰袋。
2.8 线性范围和检出限 分别准确吸取一定体积的8种氨基甲酸酯类农药的标准储备液配制成10.0mg/L工作液,用甲醇+水(4:6,V:V)逐级稀释成浓度为0.010、0.020、0.050、0.10、0.50、1.0mg/L一系列混合标准溶液,以质量浓度为横坐标(x),荧光响应信号的峰面积为纵坐标(y)进行线性拟合,结果(表2),所拟合的直线线性相关系数r为0.999 3~0.999 9,线性很好。为计算仪器信噪比(Signal-to-noise ratio,S/N),在目标物最低质量浓度附近添加了一系列已知浓度的双份样品,测定其响应信号,用S/N=3计算方法的检出限为2~5μg/kg。

表2 8种氨基甲酸酯类农药的标准曲线、线性范围、检出限
2.9 回收率、精密度及定量限 选取筛选后的柠檬、春见、爱媛空白基质,添加0.010、0.020、0.50mg/kg 3个浓度水平混合标准溶液,每个试样做3个平行质控样品,进行加标回收率试验。为模拟实样检测,向样品中加入农药后,轻微涡旋振荡离心管,密封过夜,次日按照1.3~1.7节条件进行检测,分别计算回收率和精密度。柑橘空白基质加标色谱图(图4),实验结果(表3),试样加标回收率均介于82.3%~105.1%,相对标准偏差(Relative standard deviation,RSD)为2.7%~6.2%,回收率和精密度均符合“实验室质量控制规范 食品理化检测”的分析要求[18],满足实样检测要求。经加标回收率试验验证,确定方法的定量限为0.010mg/kg。
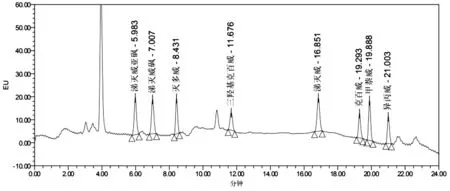
图4 柑橘空白基质加标色谱图(0.010mg/kg)
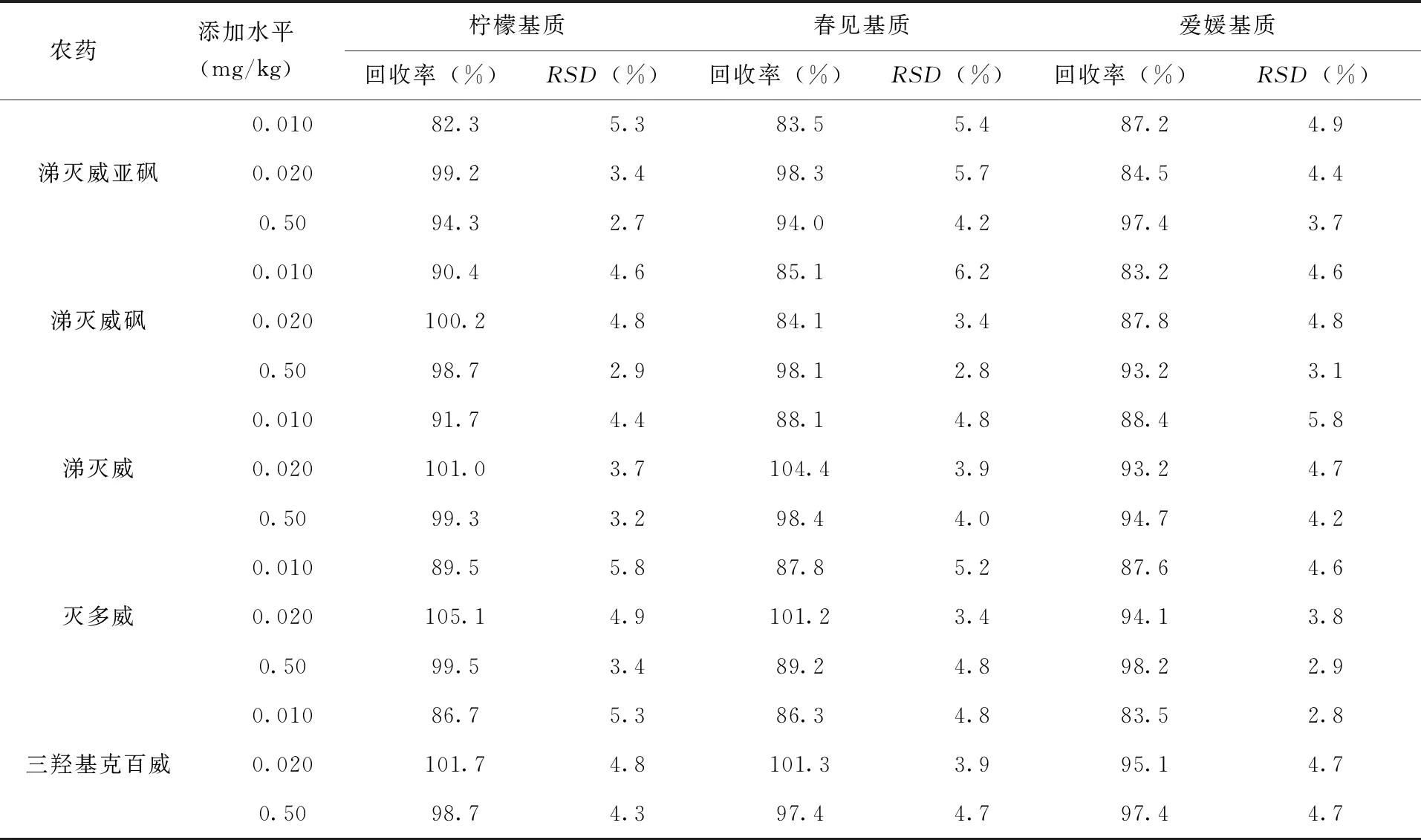
表3 柑橘加标回收率和精密度结果(n=3)
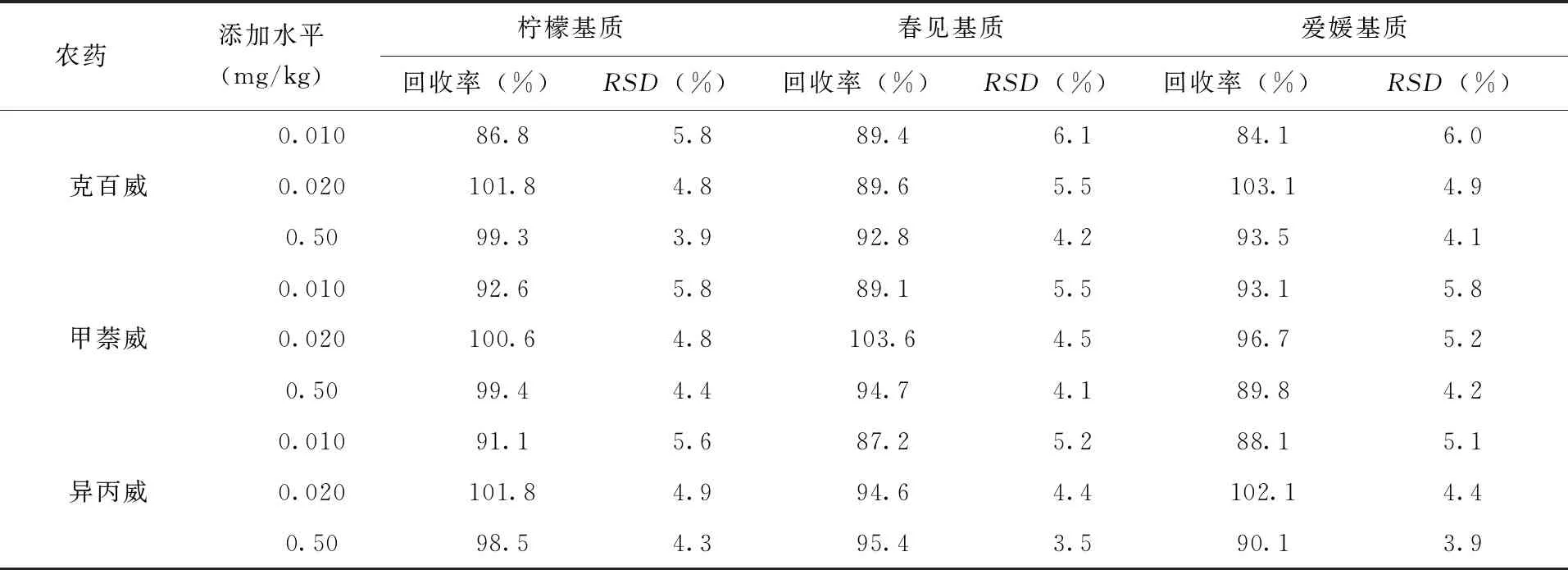
续表
2.10 实样检测 应用该方法对40个柑橘样品进行筛查检测,有2个批次样品检出克百威,其他7种农药组分均未检出。为进一步确认检测结果,用液相色谱—串联质谱法GB 23200.121-2021[19]对阳性样品进行了对比实验,结果(表4)。两种方法测定的样品含量基本一致,相对误差均<7%。查限量标准[20],克百威在柑橘类水果中的最大允许残留限量值为0.02mg/kg,第2个柠檬样品超标,后续跟进调查发现,该种植户使用了农药丁硫克百威,丁硫克百威易代谢生成克百威[21],导致样品中克百威残留超标。

表4 方法对比实验结果(n=3)
3 结论
建立了高效液相色谱柱后衍生荧光法检测柑橘中8种氨基甲酸酯类农药残留的分析方法,样品经乙腈提取、SPE-NH2固相净化,利用柱后光化学荧光衍生系统极大地提高了方法的选择性、灵敏度和抗干扰能力。该方法具有分离效果好、选择性高、通用性强及检出限低等优点,回收率、精密度和定量限均能满足农药残留分析的要求,适宜柑橘中多种氨基甲酸酯类农药的同时检测,也可为基层检测机构提供技术参考和借鉴。
猜你喜欢
煤化工(2022年3期)2022-07-08 07:24:42
毛纺科技(2021年8期)2021-10-14 06:51:16
现代仪器与医疗(2021年1期)2021-06-09 05:53:38
中国有色金属学报(2018年2期)2018-03-26 07:58:48
中国资源综合利用(2017年4期)2018-01-22 02:46:54
现代检验医学杂志(2016年1期)2016-11-12 13:19:54
中国资源综合利用(2016年10期)2016-01-22 08:36:09
中国塑料(2015年7期)2015-10-14 01:02:37
食品工业科技(2014年11期)2014-03-11 18:16:27
天津化工(2010年5期)2010-09-18 02:55:58
