
Engineered Biomimetic Platelet Membrane-Coated Nanoparticles Block Staphylococcus aureus Cytotoxicity and Protect Against Lethal Systemic Infection
2022-01-26 06:39JwaKyungKimSatoshiUhiyamaHuaGongAlexanraStreamLiangfangZhangVitorNizet
Engineering 2021年8期
Jwa-Kyung Kim,Satoshi Uhiyama,Hua Gong,Alexanra Stream,Liangfang Zhang,e,Vitor Nizet,f,*
a Division of Nephrology,Department of Internal Medicine &Kidney Research Institute,Hallym University Sacred Heart Hospital,Anyang 14068,Republic of Korea
b Department of Clinical Immunology,Hallym University Sacred Heart Hospital,Anyang 14068,Republic of Korea
c Division of Host–Microbe Systems and Therapeutics,Department of Pediatrics,University of California San Diego,La Jolla,CA 92093,USA
d Department of NanoEngineering,University of California San Diego,La Jolla,CA 92093,USA
e Moores Cancer Center,University of California San Diego Health,La Jolla,CA 92037,USA
f Skaggs School of Pharmacy and Pharmaceutical Sciences,University of California San Diego,La Jolla,CA 92093,USA
ABSTRACT Staphylococcus aureus(S.aureus)is a leading human pathogen capable of producing severe invasive infections such as bacteremia,sepsis,and endocarditis with high morbidity and mortality,exacerbated by the increasingly widespread antibiotic resistance exemplified by methicillin-resistant strains(MRSA).S.aureus pathogenesis is fueled by the secretion of toxins—such as the membrane-damaging pore-forming αtoxin,which have diverse cellular targets including the epithelium,endothelium,leukocytes,and platelets.Here,we examine the use of human platelet membrane-coated nanoparticles(PNPs)as a biomimetic decoy strategy to neutralize S.aureus toxins and preserve host cell defense functions.The PNPs blocked platelet damage induced by S.aureus secreted toxins,thereby supporting platelet activation and bactericidal activity.Likewise,the PNPs blocked macrophage damage induced by S.aureus secreted toxins,thus supporting macrophage oxidative burst,nitric oxide production,and bactericidal activity,and diminishing MRSA-induced neutrophil extracellular trap release.In a mouse model of MRSA systemic infection,PNP administration reduced bacterial counts in the blood and protected against mortality.Taken together,the results from the present work provide a proof of principle of the therapeutic benefit of PNPs in toxin neutralization,cytoprotection,and increased host resistance to invasive S.aureus infection.
Keywords:Nanoparticle Nanosponge Platelet Staphylococcus aureus Bacterial toxins Sepsis
1.Introduction
Platelets are abundant,small anucleate cell fragments in the blood circulation.The classical function of platelets involves the regulation of blood clotting and vascular integrity.However,emerging evidence has revealed a role of platelets as sentinel effector cells during infectious diseases[1–3].Innate immune responses to invading pathogens are significantly influenced by crosstalk with platelets,as the latter can sense and react to danger signals and guide leukocytes to sites of injury,inflammation,or pathogen invasion[4–7].
Platelets have multiple identified roles relevant to host defense[1,2,8,9].They can directly kill microbes by the release of antimicrobial peptides including defensins[10],cathelicidins[11],thrombocidins[12],and kinocidins[13].Platelets can also aggregate to entrap microbes and restrict pathogen spread[14].Through various different mechanisms,platelets regulate the release of a variety of intracellular mediators that are stored in granules[3].These molecules can induce inflammation and influence the recruitment and activity of effector cells of the immune system directly or indirectly[15].The mechanisms governing platelet–leukocyte and platelet–microbe interactions are complex,reflecting the diversity of platelet receptors such as complement receptors[16],Fc-γ receptor IIa[17],Toll-like receptors(TLRs)[18],glycoprotein(GP)IIb–IIIa[19],and GPIb[20].In sum,platelets are more than just clotting agents;they are critical players in the fine equilibrium of host immune and inflammatory responses.
Staphylococcus aureus(S.aureus),including methicillin-resistant strains(MRSA),is a leading opportunistic Gram-positive bacterial pathogen that produces a wide array of diseases,including invasive bloodstream infections,sepsis,and endocarditis[21,22].DeepseatedS.aureusinfections are usually accompanied by clear immune dysregulation,provoked in part by an array of secreted toxin virulence factors,including α-toxin.S.aureustoxins can engage cognate surface receptors on host cells and compromise their integrity and function by the formation of membrane pores,disruption of signal transduction pathways,or activation of enzymes that degrade host molecules[23,24].As secreted toxins play important roles in drivingS.aureusdisease pathogenesis,methods to remove or counteract toxins have become a potential therapeutic target to improve patient outcomes.In this regard,targeted antibody or nanomedicine approaches for toxin neutralization have gained attention[25–27].One such approach for biodetoxification involves natural cell-membrane-coated nanoparticles that function through biomimicry[28,29].Their unique structure allows these nanoparticles to function as decoys to absorb bacterial membrane toxin factors nonspecifically like a sponge,thereby neutralizing the factors cytolytic activity regardless of their precise molecular architecture[30].For example,red blood cell(RBC)membrane-coated ‘‘nanosponges” developed by coating polymeric nanoparticles with natural erythrocyte membranes protected mice against lethal intoxication with purified αtoxin protein[31]and reduced lesion size in murine models of MRSA or group AStreptococcusskin infection[32,33].In a mouse model ofEscherichia colisepsis,macrophage membrane-coated nanosponges bound bacterial lipopolysaccharide(LPS)and sequestered proinflammatory cytokines,thereby reducing bacterial spread and conferring protection against mortality in the treated mice[34].
Platelets have been shown to contribute to host resistance against invasiveS.aureusinfection.Antibody-mediated platelet depletion in mice impairedS.aureusclearance,as evidenced by higher bacterial burden in kidneys,more exaggerated cytokine responses,and decreased survival compared with control mice[35].Another study indicated that platelets enhanced the uptake and intracellular killing ofS.aureusby peritoneal macrophages,perhaps through a mechanism dependent on platelet granuleassociated β1-defensin[8].Platelets are an important target ofS.aureusα-toxin,as they express a disintegrin and metalloproteinase domain-containing protein 10(ADAM10),the identified α-toxin receptor,on their surface membrane[36].In mice,α-toxinmediated platelet damage and aggregation contribute to liver injury inS.aureussepsis[37].
As platelets are both important in the defense againstS.aureusand the target of the pathogen’s membrane toxins,we hypothesized that biodegradable polymeric nanoparticle cores coated with biomimetic human platelet membranes,or platelet membranecoated nanoparticles(PNPs),could be used to fortify plateletmediated defense against the pathogen.The present work provides a proof of principle of the therapeutic benefit of PNPs in toxin neutralization,cytoprotection,and increased host resistance to invasiveS.aureusinfection.
2.Materials and methods
2.1.Platelet membrane derivation
Blood-bank-approved human O-negative(universal donor)platelet-rich plasma(PRP)stored in standard acid-citratedextrose(ACD)was obtained within 24–48 h of its expiration for clinical use from the San Diego Blood Bank;this PRP serves as a source of fully functional platelet membranes,based on our prior investigations[38].Phosphate-buffered saline(PBS)solution with 50 mmol·L–1ethylenediaminetetraacetic acid(EDTA;Thermo Fisher Scientific,USA)and 300 μL protease inhibitor(PI;Thermo Fisher Scientific,USA)was added to the PRP preparation to restrict platelet activation.Platelets were centrifuged at 4000 revolutions per minute(rpm)for 15 min at room temperature;the supernatant was then discarded,and the pelleted platelets were resuspended in PBS + 1 mmol·L–1EDTA and PI tablets.
2.2.Platelet nanosponge preparation
Aliquots(1.2 mL)of platelet preparation(~3 × 109cells)were used to coat the 1 mg of poly(lactic-co-glycolic acid)(PLGA)core.Platelet membrane suspensions were derived through three cycles of freeze-thawing:The aliquots were first frozen at -80 °C,then thawed to room temperature,and then centrifuged at 8000 relative centrifugal force(rcf)for 7 min for pelleting.Finally,they were resuspended in water and quantified via the Pierce BCA protein assay kit(Thermo Fisher Scientific,USA).PNPs were derived in two stages:First,approximately 80 nm polymeric cores were prepared by nanoprecipitation using 0.67 dL·g-1carboxyl-terminated 50:50 PLGA(LACTEL absorbable polymers)dissolved in acetone at 10 mg·mL-1;1 mL of the dissolved PLGA was quickly added to 3 mL of water,and the open mixture was stirred for 12 h to evaporate the acetone to a final nanoparticle concentration of 2.5 mg·mL-1.Second,the platelet membrane preparations were combined with the nanoparticle cores at a 1:1 ratio(membrane protein to polymer weight).Platelet membrane vesicles were dispersed and fused with the PLGA particles via sonication using a frequency of 42 kHz and a power of 100 W for 5 min to achieve membrane coating.
2.3.Analysis of PNP size distribution and coating
A 1:1 membrane protein-to-polymer weight ratio yielded particles slightly larger than the PLGA core with a surface zeta potential approaching that of the platelet membrane-derived vesicles,confirming successful membrane coating.Indeed,the coating enhanced the colloidal stability of the PLGA cores,which are prone to aggregate under physiological salt concentrations.The PNPs in this study were analyzed by dynamic light scattering(Zetasizer Nano ZS ZEN 3600;Malvern Panalytical Ltd.,UK)in triplicate for size and consistency.For transmission electron microscopy(TEM),the PNPs were laid on a 400 mesh carbon-coated copper grid(Electron Microscopy Sciences,USA)stained with 1% uranyl acetate(EM Sciences,USA),and studied under a Zeiss Libra 120 PLUS energy filter transmission electron microscope(EF-TEM,Germany).
2.4.Bacterial strains and α-toxin
MRSA strain USA300/TCH1516 and its isogenic human leukocyte antigen(HLA)mutant(Sun BioRxiv,USA)were used in this study.Strains were propagated in Todd–Hewitt broth(THB;Thermo Fisher Scientific,USA)at 37 °C to mid-logarithmic phase(optical density 600 nm(OD600)= 0.4),pelleted by centrifugation at 4000 rpm for 10 min,washed once,and then resuspended to the desired dilution in PBS.Bacterial inocula-confirmed dilution plating was performed for colony-forming units(CFU).Recombinant α-toxin was purchased from Sigma-Aldrich(#H9395;USA).
2.5.Platelet isolation
Human venous blood was collected by simple phlebotomy from healthy human donors under informed consent and anticoagulated with ACD(1:6 v/v;Sigma-Aldrich,USA).PRP was prepared from blood centrifuged at 1000 rpm for 10 min with no brake,using only the upper two thirds of the PRP fractions to avoid leukocyte contamination.Platelets were isolated from PRP by centrifugation for 10 min at 1500 rpm,and then resuspended in serum-free Roswell Park Memorial Institute(RPMI)1640 media(Thermo Fisher Scientific,USA)at room temperature.
2.6.Platelet cytotoxicity assay
Human platelets(1 × 107per well)pretreated with PNPs(1 mg·mL-1)or vehicle control were placed at room temperature for 30 min,and then exposed to 3 μL supernatant of MRSA culture for 1 h.Samples were centrifuged for 5 min at 500g(g=9.8 m·s-2),and lactate dehydrogenase(LDH)released from platelets into the media was determined using the Promega assay.
2.7.Platelet bactericidal assay
To evaluate the bacterial killing by isolated platelets,isolated platelets were first pretreated with 1.0 mg·mL-1of PNPs or vehicle control for 30 min at room temperature,and then infected for 1 h with 10 μL of MRSA at multiplicity of infection(MOI)=0.1 bacteria per platelet.For CFU enumeration and to calculate the percent of MRSA killing in comparison with the original inoculum,the dilution plates were sonicated by Sonic Dismembrator 550(Thermo Fisher Scientific,USA)for 3 s.
2.8.Platelet activation assay
MRSA supernatants were collected by the centrifugation(4000 rpm for 15 min)of bacterial culture grown overnight in THB media at 37 °C.Next,1 × 107platelets were treated with 1.25–2.5 μL MRSA supernatant premixed witsupernatant of MRSA cultureh PNPs or vehicle control.After incubation at 37 °C,the samples were stained with phycoerythrin(PE)anti-human CD62p(P-selectin)antibody(Biolegend,USA)for 20 min at room temperature,and then diluted in 1 mL PBS.The expression of Pselectin was measured by FACSCalibur flow cytometry(BD Biosciences,USA)and analyzed using FlowJo v10.2 software(Becton,Dickinson and Company,USA).Human platelets and PNPs were separated by size gating;the human platelet population was then analyzed for mean fluorescence of PE.
2.9.Macrophage preparation and cell viability assays
Human myeloid leukemia monocytes(THP-1)(American Type Culture Collection(ATCC),USA)were cultured in RPMI + 10% fetal bovine serum(FBS).The THP-1 were differentiated into macrophages with 25 nmol·L–1phorbol 12-myristate 13-acetate(PMA;Sigma-Aldrich,USA)for 48 h followed by a 24 h cooldown period in RPMI+10%FBS.For THP-1 cytotoxicity,5×105THP-1 were plated in each well and pretreated with 1 mg·mL-1of PNPs or vehicle control for 30 min at room temperature,and then exposed to a range of MRSA supernatant doses(1.25–10.00 μL)for 1 h at 37°C.The viability of THP-1 and THP-1 differentiated macrophages was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte-trazolium bromide(MTT)cell proliferation assay kit(ab211091;Abcam plc.,USA),which quantifies adenosine triphosphate(ATP)conversion of MTT to formazan as read by absorbance at 590 nm.
2.10.Macrophage bactericidal assays
For THP-1 differentiated macrophage bactericidal assays,differentiated macrophages were treated with 1 mg·mL-1PNPs or vehicle control for 30 min,and then infected with MRSA at MOI = 1.0 bacteria per cell.Cells were lysed using Triton X-100(0.025%;Sigma-Aldrich,USA),and serially diluted for CFU enumeration and the calculation of percent MRSA killing in comparison with the original inoculum.
2.11.Macrophage oxidative burst and nitric oxide production assays
For oxidative burst assays,THP-1 differentiated macrophages were loaded with 25 μmol·L–12,7-dichlorofluorescein diacetate(DCFH-DA;Thermo Fisher Scientific,USA)in Hank’s balanced salt solution(HBSS;Mediatech,USA)lacking Ca2+and Mg2+,and rotated at room temperature for 30 min.Macrophages were then infected with MRSA(MOI = 1.0 bacteria per cell)with or without PNPs,and incubated at 37 °C.Every 15–30 min,the fluorescence intensity at 485 nm excitation/520 nm emission was compared by SpectraMax M3(Molecular Devices,USA).For quantifying the nitrite production in THP-1 differentiated macrophages under the same exposure conditions,Greiss reagent(Promega,USA)was used according to the manufacturer’s protocol.
2.12.Human neutrophil extracellular trap staining and quantification
Neutrophils were isolated from blood collected from healthy adult human donors who had given informed consent,using PolymorphPrep(Progen Biotechnik GmbH,Germany)according to the manufacturer’s instructions.Next,5×105neutrophils were placed in the wells of a 24-well plate,stimulated with vehicle control,25 nmol·L–1PMA,MRSA alone,or MRSA premixed with PNPs for 20 min(MOI = 10 bacteria per neutrophil),and incubated for 3 h at 37°C.For visualization,cells fixed in 4%paraformaldehyde were stained with an antibody against myeloperoxidase(MPO)(1:300;Calbiochem,USA)in PBS + 2% bovine serum albumin(BSA;Sigma-Aldrich,USA)for 1 h,then placed in Alexa Fluor 488 goat anti-rabbit(1:500;Life Technologies,USA)for 45 min,and finally counterstained with 1 μmol·L–1Hoechst-3342-trihydrochloride diluted in 2% PBS–BSA for 10 min before imaging was carried out on a fluorescence microscope.In parallel,neutrophil extracellular traps(NETs)were quantified using the Quant-iTTM PicoGreen dsDNA assay kit(Invitrogen,USA)on wells in which micrococcal nuclease solution was added to release the DNA of the NETs into the supernatant;500 mmol·L–1EDTA was added to the solution to stop the micrococcal nuclease reaction.PicoGreen solution was prepared according to the manufacturer’s instructions and incubated for 5 min;fluorescence signals were then measured with filter settings of 480 nm excitation/520 nm emission.
2.13.Cytokine quantification assays
Cytokines interleukin(IL)-8 and IL-1β were quantified from infected THP-1 differentiated macrophage cell supernatants(4,8,and 24 h post-infection)using enzyme-linked immunosorbent assay(ELISA)kits(R&D systems,USA)according to the manufacturer’s protocol.Experiments were conducted in triplicate or quadruplicate.
2.14.In vivo murine S.aureus infection experiments
For survival studies,MRSA cultures were grown in THB to midlog phase and washed once in PBS.Next,3 × 108CFU was intraperitoneally(i.p.)injected in outbred,10–12 week-old,CD1 mice(Charles River,USA).For the PNP treatment group,100 μL of a 5 mg·mL-1of PNPs was injected intravenously(i.v.)twice,immediately after MRSA infection and 3 h after the first injection.Survival was monitored daily for 6 d.In a separate challenge experiment using the same MRSA challenge dose and PNP treatment protocol,mice were sacrificed at 6 h for the determination of bacterial CFU units and the quantification of tumor necrosis factor(TNF)and IL-6 in serum and spleen.
2.15.Statistical analyses
All studies were conducted in duplicate or triplicate and repeated independently at least twice.All data are graphed as means with standard error of the mean(SEM)or standard deviation(SD).Statistical evaluation was done by one-way analysis of variance(ANOVA)or unpaired two-tailed Student’st-test(Graph Pad Prism 5.03)(*:P<0.05;**:P<0.01;***:P<0.001).
2.16.Ethical approval
Animal studies were conducted in accordance with protocols approved by the UC San Diego Institutional Animal Care and Use Committee(IACUC;protocol S00227M);all efforts were made to reduce animal numbers and minimize suffering.Blood for platelet isolation was obtained via venipuncture from healthy volunteers under written informed consent approved by the UC San Diego Human Research Protection Program.
3.Results
3.1.Preparation and analysis of PNPs
In this study,our goal was to construct PNPs composed of a PLGA polymer core wrapped in natural platelet bilayer membranes.The platelet membrane shell provides a faithful mimicry of the parent platelet surface and can thus absorb the toxin virulence factors of the MRSA of diverse molecular structures,with the aim of reducing platelet toxicity and preserving platelet defense function against MRSA infection(Fig.1(a)).Cell membranes from recently expired(<24–48 h)blood-bank-approved platelet concentrates from hypotonic lysis,mechanical disruption,and differential centrifugation have been found to retain membrane composition and attendant functions[38].EDTA was used as the anticoagulant to chelate divalent cations including calcium and avoid activating coagulation processes while stabilizing the platelets[38].Furthermore,a PI was added to block platelet aggregation[38].A sonication procedure yielded membrane vesicles for fusion onto PLGA cores to create the final PNPs.The inner polymeric core stabilizes the outer membrane from collapsing and fusing with other membranes,optimizingin vitroandin vivostability.After membrane coating,the PNP diameter increased from(88.4 ± 5.6)to(120.0 ± 4.8)nm as assessed by dynamic light scattering,reflecting the encapsulation of the polymeric cores with a cell membrane bilayer(Figs.1(b)and(c)).TEM clearly showed PLGA polymer cores uniformly coated by a unilamellar membrane,indicating successful PNP formation(Fig.1(d)).
3.2.PNPs prevent human platelet damage and dysfunction induced by MRSA supernatants

Fig.1.Formulation and analysis of PNPs.(a)Model showing our rationale of applying PNPs to modulate immune cells,pathogen binding,and control in vivo therapeutic efficacy.(b,c)Dynamic light scattering to evaluate hydrodynamic size(diameter in nm)of the PLGA polymeric cores before and after platelet membrane coating.(d)TEM images of the derived PNPs using uranyl acetate counterstain.
Bacterial toxins includingS.aureusα-toxin can damage and inhibit the functions of host cells[35,39].We first investigated whether PNPs could protect intact human platelets against the cytotoxic effect of MRSA supernatants.When human platelets were incubated with MRSA supernatant for 1 h,a significant LDH release reflecting platelet damage was seen;however,this was significantly diminished in the presence of PNPs:The LDH release was 0.52±0.13(optical density(OD)value)in MRSA supernatant alone,in comparison with 0.27 ± 0.08(OD value)with PNP treatment(P= 0.002)(Fig.2(a)).The activation of platelets associated with the release of antimicrobial peptide-rich α granules[2]can be quantified by the surface expression of P-selectin.Upon exposure of human platelets to MRSA supernatants,we found that coincubation with PNPs significantly reduced the time to P-selectin expression and significantly increased the intensity of its expression(Figs.2(b)and(c)),indicating that PNPs could help to preserve this key platelet response function.To examine whether platelet cytoprotection and functional activation improved innate immune activity,MRSA killing by platelets was assessed in the presence or absence of PNPs.A significant enhancement of platelet MRSA killing was seen upon PNP treatment(Figs.2(d)and(e)),suggesting that the protective effect of PNPs on MRSA-induced platelet injury might improve the host defense function.
3.3.PNPs prevent human macrophage damage and dysfunction induced by MRSA supernatants

Fig.2.PNPs prevented human platelet damage and dysfunction induced by MRSA supernatants(S/N).(a)PNP reduced MRSA supernatant-induced platelet cytotoxicity as measured by LDH release with OD value;the cytotoxicity was 0.52±0.13 in MRSA supernatant alone,0.27±0.08 with PNP treatment(P=0.002).(b,c)P-selectin expression on platelets exposed to MRSA supernatant in the presence or absence of PNPs.PNP treatment led to a striking increase of P-selectin expression beginning at the early time point(10 min).(d,e)Increased platelet viability upon PNP treatment is accompanied by improved MRSA killing.**: P <0.01;***: P <0.001.

Fig.3.PNPs prevented macrophage damage and dysfunction induced by MRSA supernatants(S/N).(a)LDH cytotoxicity test and(b)MTT assay indicated that PNP treatment showed about 75% reduction of THP-1 cytotoxicity.(c)PNP also improved the viability of THP-1 differentiated macrophages after MRSA supernatant treatment.(d)Pre-treatment of THP-1 differentiated macrophages with PNP showed higher bactericidal efficiency regardless of bacterial load and incubation time.*: P <0.05;**: P <0.01;***: P <0.001.
The effect of MRSA supernatants on the viability of THP-1 was examined with or without preincubation of the MRSA supernatant with PNPs for 30 min beforehand.As shown in Fig.3(a),a nearly 75% reduction in LDH release was observed in the presence of PNPs.Cell viability was further assessed by MTT assay,which showed that the viability of THP-1 after 1 h treatment was markedly increased when the MRSA supernatant was premixed with PNPs,in comparison with the control(Fig.3(b)).LDH release was also measured in the THP-1 differentiated macrophages,and revealed a significant protective effect of PNPs(Fig.3(c)).Finally,the THP-1 differentiated macrophages were infected with live MRSA in the presence or absence of PNPs,showing that the nanosponge treatment significantly increased macrophage killing of the pathogen(Fig.3(d)).
3.4.PNP protection against cytotoxicity preserves macrophage and neutrophil effector responses
We further examined the effect of PNPs on certain key macrophage functions involved in the antibacterial response.The generation of reactive oxygen species(ROS),known as oxidative burst,is a key element of macrophage antibacterial defense,as evidenced by the high rate ofS.aureusinfections in nicotinamide adenine dinucleotide phosphate(NADPH)oxidase deficiency(chronic granulomatous disease)patients with impaired oxidative burst function[40].PNP treatment increased THP-1 differentiated macrophage ROS production in response to MRSA exposure(Fig.4(a)).Nitric oxide generation by inducible nitric oxide synthase(iNOS)also contributes to antibacterial defense,as corroborated by more severeS.aureusinfection in iNOS-deficient mice[41].PNP treatment likewise boosted THP-1 differentiated macrophage nitric oxide production in response to MRSA exposure(Fig.4(b)).S.aureusinfection induces macrophage pyroptosis,a cell death program that is dependent on inflammasome activation and associated with IL-1β release[42].The generation of IL-1β was modestly reduced at the early time point(Fig.4(c));this was coincident with reduced LDH release in cytotoxicity assays(Fig.3(a)),indicating that pyroptosis is one element of the macrophage response toS.aureuschallenge,and that the degree of associated cytotoxicity and cytokine release can be partially mitigated by PNP toxin absorption.Platelets express the IL-1β receptor(IL-1R)[43],so sequestration of the IL-1β cytokine by PNP may also contribute to the observed difference.Finally,S.aureustoxins[44,45]and activated platelets[46]also elicit neutrophil extracellular traps(NETs),with potential proinflammatory and procoagulant effects that may aggravate sepsis[47]or endocarditis[48].PNP treatment significantly inhibited MRSA-induced NET formation by human neutrophilsin vitro,as visualized by immunostaining and measured by PicoGreen quantification of released DNA(Figs.4(d)and(e)).
3.5.PNPs improve survival in a murine MRSA systemic infection model
Ourin vitrostudies described above show that PNPs blocked MRSA-induced platelet and macrophage cytotoxicity,enhancing the antibacterial effectiveness of both cell types.These findings suggest a potential therapeutic benefit of PNPs againstS.aureusinfectionin vivo.In prior rodent i.v.injection studies for pharmacokinetic and biodistribution assessment,more than 90% of PNPs were distributed to tissues within 30 min,most prominently in the liver and spleen[38].Importantly,abnormal blood coagulation has not been seen in prior uses of PNPs in murine models of noninfectious indications,including immune thrombocytopenia[49]and atherosclerosis[50].We challenged groups of eight mice with 3 × 108CFU MRSA in 100 μL volume i.p.to induce systemic infection.For the PNP treatment group,we injected 5 mg·mL-1PNPs directly into the bloodstream IV immediately after MRSA infection,and again 3 h after the first injection(Fig.5(a)).The goal of the two doses was to provide a modest extended therapeutic coverage window,seeking to mitigate pathogen-mediated toxic damage,prevent the propagation of cytokine storm,and prevent early death from sepsis.The survival of mice treated with PNP was significantly improved,with 37.5% of mice surviving for 5 d despite the absence of antibiotic therapy,whereas all mice in the control group died within the first 48 h(Fig.5(b)).In a separate experiment using the same challenge and treatment conditions,we collected blood at 6 h post-MRSA challenge for the determination of bacterial CFU and serum cytokine levels.The two-dose PNP treatment was associated with a significant(P= 0.036)reduction of MRSA CFU in blood(Figs.5(c)and(d)),and a slight trend toward reduced counts in the spleen that did not reach statistical significance.Serum IL-6 levels in response to the MRSA challenge were also reduced in the PNP-treated group compared with the control group(Fig.5(e)).
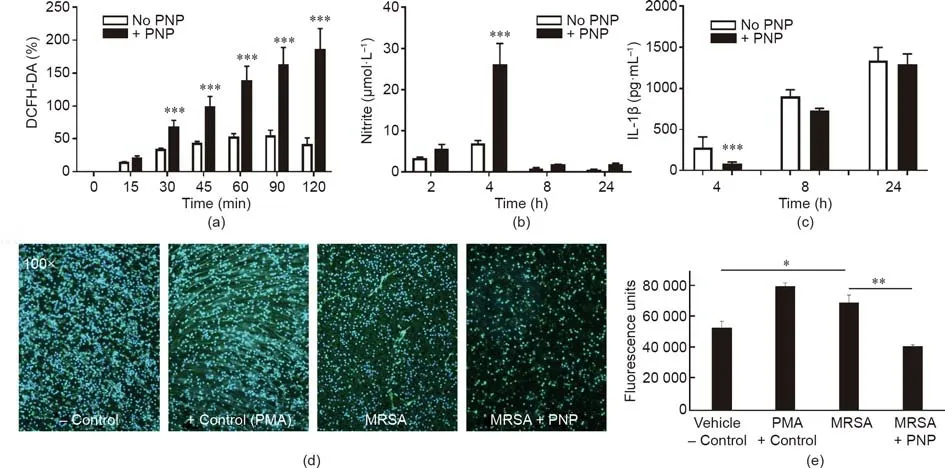
Fig.4.Effect of PNPs on activation of macrophage and neutrophil bactericidal mechanisms.(a)PNP treatment increases macrophage oxidative burst,as measured by DCFHDA assay for superoxide production.(b)Increased nitrite production reflecting nitric oxide production was observed in PNP-treated macrophages.(c)Reduced cytokine IL-1β production at the earliest(4 h)time point in PNP-treated macrophages.(d)Immunostaining of human NETs elicited by MRSA in the presence or absence of PNP treatment;PMA serves as a positive control.(e)Quantification of NETs by PicoGreen assay.*: P <0.05;**: P <0.01;***: P <0.001.

Fig.5.PNPs improve survival in a murine MRSA systemic infection model.(a)Schematic diagram of the setup of in vivo experiments.(b)Survival rates of mice over 144 h following an i.p.injection of MRSA(3 × 108 CFU per mouse).100 μg PNP at 5 mg·mL-1 was injected twice,at 0 and 3 h after bacterial inoculation(n = 8 in each group).Treatment with PNPs provided a significant survival benefit.(c–e)Enhanced bacterial clearance in blood,spleen,and serum during PNP treatment.
4.Discussion
In this study,we provided a proof of principle of the therapeutic potential of PNPs in systemic MRSA infection.This benefit is likely multifactorial,and may include:①reduction of toxin-associated platelet damage,which allows for more rapid and effective deployment of platelet antimicrobial activity;②protection of phagocytic cells such as macrophages from MRSA cytolytic injury,allowing them to more efficiently deploy ROS and NO and achieve effective bacterial killing;and ③sequestration of certain bacterial toxins(e.g.,pore-forming toxins)in the platelet membrane.Also,since platelets express TLRs(e.g.,TLR2)and cytokine receptors(e.g.,IL-1R),they could scavenge proinflammatory bacterial cell wall components and cytokines to modulate the hyperinflammation of sepsis.The latter mechanisms would parallel thein vivotherapeutic effects of macrophage membrane-coated nanoparticles,which conferred protection against lethality in murine models of LPS endotoxemia andEscherichia colisepsis[34].While prior work showed that RBC nanosponges protected mice from lethal challenge with whole secreted protein preparations fromS.aureussupernatants[31],the present work with PNPs is the first report of protection by biomimetic membrane-coated nanoparticles against systemic infection with liveS.aureus.
A recent study suggests that sepsis contributes as much as 19.7% to human mortality worldwide[51],and there remains no drug specifically approved for the treatment of sepsis[52].Expanding numbers of antibiotic-resistant bacterial strains such as MRSA further complicate effective sepsis therapeutics[53].Against this background,the use of PNPs or other cell-membrane-coated nanosponges in sepsis provides a more ‘‘universal” approach to the absorption and neutralization of bacterial toxins,in contrast to monoclonal antibodies or other platforms targeting the particular molecular structure of an individual toxin or host inflammatory factor.Other findings that might support an effective therapeutic profile of PNPs include evidence that they reduce macrophage cellular uptake,do not induce human complement activation in autologous plasma,selectively bind to damaged vascular endothelial cells from human and rodents,and likewise bind to plateletadherent pathogens[31],potentially favoring their accumulation at common sites of endovascularS.aureusinfection.
In sum,our data suggests that PNPs may protect platelets during systemic MRSA infection by absorbing circulating toxins so that platelets sustain less damage,survive longer,activate more quickly,and have stronger antibacterial effects.PNPs might also protect and support the innate immune function of macrophages or other phagocytic cell types.This biomimetic detoxification strategy merits further exploration as an adjunctive therapy to improve clinicals in patients with MRSA bloodstream,thereby expanding the current approach to clinical management.
5.Limitations of this study
Murine studies of systemic MRSA infection cannot fully recapitulate the tempo and kinetics of human infection,as mice are relatively resistant to the pathogen and very high challenge doses are required to cause organ dissemination and mortality risk.Furthermore,in patients with presumed sepsis,rapid clinical intervention is required—often before microbiological confirmation of the pathogen is established—and patients may have one or more comorbidities that increase the risk of an adverse outcome.Nanosponge therapeutics are thus envisioned as an addition to the standard of care.
Acknowledgements
This work was supported by National Institutes of Health grants HL125352 and U01AI124316(VN).
Compliance with ethics guidelines
Jwa-Kyung Kim,Satoshi Uchiyama,Hua Gong,Alexandra Stream,Liangfang Zhang,and Victor Nizet declare that they have no conflict of interest or financial conflicts to disclose.

- Engineering的其它文章
- Selective Laser Melting under Variable Ambient Pressure:A Mesoscopic Model and Transport Phenomena
- Programmable Adaptive Security Scanning for Networked Microgrids
- Flexibility Prediction of Aggregated Electric Vehicles and Domestic Hot Water Systems in Smart Grids
- An Overview of Metal–Organic Frameworks for Green Chemical Engineering
- Atomic Force Microscopy Measurement in the Lignosulfonate/Inorganic Silica System:From Dispersion Mechanism Study to Product Design
- Advances and Strategies for Controlling the Quality and Safety of Postharvest Fruit