
碱性水电解非贵金属析氧催化剂的研究进展
2022-01-23 04:20郭丹丹滕越迟军姚德伟俞红梅邵志刚缪春辉高强
可再生能源 2022年1期
郭丹丹,滕越,迟军,姚德伟,俞红梅,邵志刚,缪春辉,高强
(1.中国科学院大连化学物理研究所,辽宁大连 116023;2.中国科学院大学,北京 100039;3.国网安徽省电力有限公司电力科学研究院,安徽合肥 230061;4.国网浙江电力有限公司台州供电公司,浙江台州 318000)
0 引言
氢气具有能量密度高、清洁无污染等优势,被认为是“绿色”的能源载体[1]。在众多的制氢方法中,水电解制氢由于环境友好、产品纯度高以及无碳排放等优点,成为最具发展潜力的“绿色”制氢方法[2]。水电解过程涉及两个半反应,即析氢反应(HER)和析氧反应(OER)。和HER相比,OER涉及复杂的多电子-质子耦合转移步骤,导致动力学缓慢,从而限制了水电解的效率[3],[4]。
受限于质子交换膜(Proton Exchange Membrane,PEM)水电解制氢体系的强酸和高氧化电位运行工况,传统的非贵金属催化剂在此工况下不稳定,不能满足长时间稳定运行的需求。Ir基和Ru基催化剂在酸性和碱性介质中均具有良好的OER催化活性,常被用作评价OER体系的基准催化剂,但其相对较高的成本和稀缺的资源限制了其广泛应用[1],[5],[6]。
尽管目前已开发出大量适用于碱性体系的非贵金属OER催化剂,但相关催化剂的催化活性仍然较差,距离实际应用仍有一定差距。因此,在碱性体系中,研制具有催化活性好、稳定性高、成本低的非贵金属(Fe,Co,Ni和Mo等)OER催化剂仍是电解水技术中亟待解决的关键问题[1]。
本文是对近两年碱性体系中OER非贵金属催化剂活性提升策略的研究进展的总结,分别从材料掺杂类、形貌调控类、调节电子结构类和复合结构类4个方面归纳了目前非贵金属催化剂发展所面临的问题和挑战。为进一步开发具有高效、廉价和稳定特性的碱性水电解制氢非贵金属基OER催化剂提供了新的思路。
1 碱性OER催化剂的设计原则
OER是电解水过程中的关键半反应,涉及到复杂的四电子转移过程,其动力学反应缓慢,过电势较高,限制了电解水制氢的转化效率。开发高效的OER电催化剂,对加快反应进程、提高OER的转化效率至关重要。目前,活性较好的非贵金属催化剂的导电性较差,在高电流密度下运行时的过电位较高,和贵金属催化剂相比仍有较大差距。
电化学反应的电压损失主要由活性极化、欧姆极化和传质极化造成,因此,要想设计出高效的非贵金属OER电催化剂,须从这3个方面进行改进,通常要遵循以下原则:①活性好,降低活性极化,原材料含量丰富可实际应用;②比表面积大,能增加活性位点的数目;③亲水性好,避免电极材料表面生成大量气泡,从而加剧传质极化损失;④导电性好,能够在高电流密度运行时保持较低的欧姆极化损失;⑤耐腐蚀且电化学稳定性好。
2 非贵金属OER催化剂
2.1 材料掺杂类OER催化剂
大量的研究表明,在非贵金属催化剂中掺杂IIIA(B),VA(N和P等)和VIA(O,S和Se等)主族元素或过渡金属元素(Fe,Co和Ni等),可以调节OER电催化剂活性位点周围的电子结构并提高非贵金属催化剂的OER性能。
FeNi基层状双金属氢氧化物(LDHSs)是目前报道的活性较好的OER电催化剂之一。然而,通常条件下制得的LDHSs催化剂呈片状,易堆叠,制成的催化层较厚,导致传质极化较为严重。Xie X Y[2]以不锈钢材料(SS)作为金属源和基底,制备了铬单原子掺杂的超薄FeNi-LDHSs纳米片阵列自支撑电极(Cr/FeNi-LDHS-SS)(图1)。Cr的掺杂,调节了活性位点的电子结构,使Cr/FeNi-LDHS-SS电极表现出优异的OER性能,当电流密度分别为10,100 mA/cm2时,其过电势为202,242 mV。该研究以降低催化层厚度、改善催化层导电性、元素掺杂和提高催化剂本征活性的方式来实现高活性非贵金属析氧电极的制备,为设计廉价高效的自支撑电催化剂提供了一个新思路。

图1 FeNi-LDH和Cr/FeNi-LDH在OER过程中的结构构型Fig.1 Structural configurations of the OER process:FeNi-LDH and Cr/FeNi-LDH
Liu Q H[7]采用一步化学还原反应将一维Fe2B纳米线(NWs)直接沉积到三维的泡沫镍(NF)上,制备出了立体化的Fe2B纳米线网络。研究发现,相互连接的Fe2B纳米线在碱性溶液中的水电解析氧反应过程中会转化为偏硼酸盐和FeOOH覆盖的更厚、更大的Fe2B纳米线网络。在Fe2BNWs/NF的催化作用下,当电流密度为10 mA/cm2时,OER的过电位为276 mV。
此外,过渡金属氧化物(TMOs),如Co3O4,被认为是贵金属氧化物的有效替代品。Zhang S L[8]报道了铁原子掺杂的超薄Co3O4纳米片合成的空心纳米片。通过实验发现,掺杂少量铁原子的Co3O4材料可以增强OER的电催化活性,这是由于铁原子被掺杂到Co3O4的八面体中,铁元素和钴元素价态相似,金属间通过配位形成独特的通道。在Fe-Co3O4的催化作用下,当电流密度为10 mA/cm2时,OER的过电位为262 mV。
Jiang Y Y[9]制备了硼原子掺杂石墨烯负载的钴镍双金属氧化物,其中Co-Ni-Ox/BG(Co/Ni为1:1)在碱性条件下具有良好的OER电催化性能,当电流密度为10 mA/cm2时,OER的过电位为310 mV,该结果优于已报道的单金属氧化物的催化剂。这是因为硼掺杂到CoNi双金属氧化物复合材料中后,可以调控金属原子周围的电荷分布,从而对电催化起到促进作用。
Zhang Z[1]利用室温下的自发氧化还原反应来合成纳米银修饰的过渡金属氢氧化物(TMHs)复合材料OER催化剂。在碳纤维布上生长的Ag@Co(OH)x具有出色的OER活性,当电流密度为10 mA/cm2时,OER的过电位为250 mV。
以上研究表明,将非金属(如Cr或B)原子、非贵金属(如Fe)原子和少量Ag原子掺杂到非贵金属骨架中,能改性催化剂的电子结构,使之具有良好的电荷传输能力和亲水表面,从而提升OER性能。
2.2 形貌调控类OER催化剂
构建具有特殊结构的OER电催化剂可以改善电极表面的传质极化问题,也是改善水分解反应动力学的重要策略。这种方法是提高催化材料性能和优化表面界面催化反应的有效方法。
Zhang J T[10]通过模板法合成了双壳结构的Ni-Fe层状双金属氢氧化物(LDHS)纳米笼OER催化剂(图2)。这种结构具有较高的电化学活性表面积,在1M的KOH溶液中,双壳结构的Ni-Fe LDHS纳米笼催化剂展示出了较好的OER催化性能,当电流密度为20 mA/cm2时,其OER过电位为246 mV。
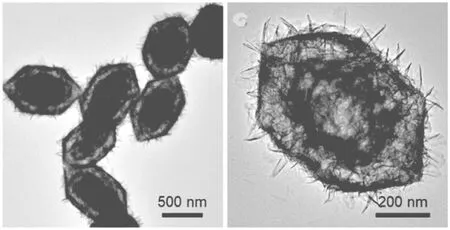
图2 双壳Ni-Fe LDHS纳米笼OER电催化剂的TEM图Fig.2 TEM images of the double-shelled Ni-Fe LDHS nanocages OER electrocatalysts
Xia J L[11]发现了一种由NiFeCr氢氧化物沉积在多孔Cu@CeO2纳米管阵列上形成的催化剂(Cu@CeO2@NFC-0.25),其中,CeO2的引入促进了氧离子向催化中心的转移,有效调节了NiFeCr氢氧化物的电子结构。NFC和CeO2界面之间的协同优化,丰富了催化剂的活性位点并促使d-f轨道耦合,促进了电子转移以及氧的快速扩散和释放;管状结构中的铜芯具有较高的导电性,从而提升了催化剂在碱性电解液中的OER活性。在Cu@CeO2@NFC-0.25的催化作用下,当电流密度为10 mA/cm2时,OER的过电位为230.8 mV。
Li Y[12]研究了一种由一维晶态磷化钴纳米线和二维无定型镍铁氢氧化物纳米片组成的具有“枝-叶”结构的催化剂(CoP@NiFe-OH),这种分层形貌结构使内、外层两组分在电解液中充分暴露,不仅提供了更多的催化活性位点,还加快了传质过程。在碱性OER性能测试中,CoP@NiFe-OH仅需220 mV的过电位即可产生20 mA/cm2的析氧电流。
Huang L[13]采用原位自解离组装方法制备了CoNi泡沫合金上负载超薄CoNi-MOF的纳米片阵列材料(CoNi-MOFNA)并将其用于OER。在碱性介质中,在该催化剂的作用下,当电流密度为10 mA/cm2时,对应的OER过电位为215 mV。在OER电化学性能测试过程中,CoNi-MOFNA表面生成的均匀的超薄羟基氧化物团簇会与MOF杂化,这不仅稳定了催化剂的空间结构,也保护了活性位点。
以上研究表明,通过设计具有特殊结构的形貌,可对催化剂进行优化,扩大催化剂的电化学有效活性面积、加快质子传输和电子转移能力并增加活性位点,从而显著提升催化剂在OER中的性能。
2.3 调节电子结构类OER催化剂
提升OER催化剂的另一种有效方法是通过阴阳离子对催化剂局部配位环境进行调控,如在催化材料中引入缺陷和空位来调整纳米材料的固有电子结构,从而提升非贵金属基OER电催化剂的性能。
Wang Y Q[14]通过甲基异氰酸盐(CH3NCS)在NiFe-LDHs的边缘和基面形成金属和氧间的多个空位,来提升NiFe-LDHs的析氧催化活性。金属和氧间的空位可以提高NiFe-LDHs的电子转移能力并调节H2O的吸附,从而提高OER的电催化性能。具有金属和氧的多个空位的催化剂(v-NiFe-LDHs)性能最佳,在其作用下,当电流密度为100 mA/cm2时,OER的过电位为230 mV。
通常来说,阳离子是过渡金属化合物电催化剂中的真正活性位点,其电催化活性受周围阴离子结构的调节。Zhao C X[15]提出了一种阴离子辅助调节的电化学反应,用于精确构建具有高电催化活性的电催化剂(图3)。该方法制备的羟基硫化物电催化剂具有特殊的电子结构,因为采用电化学辅助方法能够可控调节阴离子,使原子充分分散,从而提升了电催化剂的活性。

图3 阴离子辅助调节电化学反应的示意图Fig.3 Schematic of the anionic regulation assisted by electrochemical reaction
Kuang M[16]采用阳离子(Fe3+)交换法制备了钴-钒-铁(氧)氢氧化物(CoV-Fe0.28)纳米片。在CoV-Fe0.28中,Co,V和Fe离子间的协同作用调节了局部配位环境和电子结构。在OER电化学性能测试中,CoV-Fe0.28表现出了优异的电催化活性,当电流密度为10 mA/cm2时,OER的过电位为215 mV。
以上研究通常是通过阳离子调控、阴离子调控、缺陷工程和表面修饰等方式来调整材料表面的电子结构,从而增强材料的电学性能,这些方法都是设计出高效OER催化剂的有效途径。其中,阳离子调控能暴露更多的活性位点、优化催化剂的吸附态和电荷转移特性,而阴离子调控能调节催化剂的电子特性和增加反应位点。在材料中引入杂原子来破坏晶格的周期性使局部的电子结构发生改变,而电子结构的变化可有效改变反应中间体的吸附能力,从而提高电催化的效率。
2.4 复合结构类OER催化剂
构造复合结构是指在同一个催化剂中将不同活性组分的纳米材料复合在一起,使其产生界面效应,从而暴露出更多的活性位点,提高电荷转移能力。构造复合结构也是提升非贵金属OER催化剂活性和稳定性的有效方法。
Li X P[17]用连续两步电沉积的方法,制备了含有两种异质结构(即Ni-NiO和MoO3-NiO)的非晶态NiO纳米片(MoO3/Ni-NiO)(图4)。Ni-NiO和MoO3-NiO的异质结构降低了能量势垒,协同加速了OER催化剂的活性中心,所以MoO3/Ni-NiO复合材料具有优异的电催化性能。在OER电催化性能测试中,当电流密度为100 mA/cm2时,OER的过电位为347 mV。

图4 制备M oO3/Ni-NiO/CC催化剂的示意图Fig.4 Schematic illustration for the preparation of MoO3/Ni-NiO/CC
Yang Y[18]合成了一种富相界面的四面体NiS2/NiSe2多级笼异质结催化剂。这种异质结结构可暴露出更多的活性位点,能加速离子和气体传质,并优化界面电子结构。具有双相协同作用的四面体NiS2/NiSe2多级笼能有效地触发OER反应过程,并加速OER动力学。在OER电化学性能测试中,当电流密度为20 mA/cm2时,OER的过电位为290 mV。
Jiang J Y[19]采用一步低共熔溶剂法在碳纤维纸基底上构建了具有异质结构的二维CoNC@Co2NHS@CP催化剂。Co2N纳米片暴露的表面具有高表面能,可锚定高活性的CoNC,在碳纤维纸(CPs)外部形成二维CoNC@Co2N异质结构。这种结构能促进电子传输,同时降低催化能垒,二维非范德华纳米片的桥连特性使得CoNC纳米片和CPs之间形成了牢固且导电的连接。在OER电化学性能测试中,当电流密度为10 mA/cm2时,OER的过电位为217 mV。
Gao X R[20]设计了由Ni3N/Ni异质结构核和Ni3N壳组成的强耦合核壳结构纳米催化剂(Ni3N/Ni@Ni3N)。当OER的过电位为270 mV时,与Ni3N,Ni和商用RuO2相比,Ni3N/Ni@Ni3N的每单位有效表面积的催化电流密度分别提高了17,37和20倍。这是因为在OER中,Ni3N/Ni@Ni3N产生了一种自适应的NiOOH物种表面重构,提升了催化剂的活性。
以上研究是通过构建复合异质纳米结构,使其在杂化催化剂的纳米级界面上产生协同效应,从而有效提高非贵金属催化剂在碱性溶液中催化OER的性能。
通过对材料掺杂类、形貌调控类、调节电子结构类和复合结构类非贵金属基OER催化剂的研究,可以发现,在碱性体系中对非贵金属基OER催化剂的研究已取得了一定成效[21],非贵金属基OER催化剂在碱性水电解中具有很大的发展空间。
3 结语
本文主要介绍了4种研究方法来提升非贵金属基OER催化剂的活性和稳定性:①掺杂IIIA(B),VA(N和P等)和VIA(O,S和Se等)主族元素或过渡金属元素(Fe,Co和Ni等),调节OER电催化剂活性位点周围的电子结构来提升性能;②设计特殊的形貌结构对催化剂进行优化,扩大电化学有效活性面积、加快质子传输和电子转移能力;③在催化材料中引入缺陷和空位来调节催化剂的电子结构;④构造复合结构,将不同活性组分的纳米材料复合在一起,使其产生界面效应,从而暴露出更多的活性位点,来提高电荷转移能力和改进催化性能与稳定性。
近年来,在碱性体系中对非贵金属基OER催化剂的研究工作进展较快,但催化剂的活性和稳定性仍不能满足实际应用。在碱性OER过程中,大多数研究都是在三电极体系中进行的,而将催化剂材料用于水电解槽测试的研究,鲜有报道。此外,对于活性物种的研究仍不明确,不能确定活性物种是如何影响OER活性的。从原子水平上去确定活性中间体的种类是很有必要的,但是,现有表征手段的分辨率有限,难以确定催化剂复杂的表面结构,使得这项研究工作面临巨大挑战。
猜你喜欢
分子催化(2022年1期)2022-11-02
家庭科学·新健康(2022年7期)2022-07-13
蓄电池(2022年1期)2022-02-25
表面技术(2022年1期)2022-02-12
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26
伴侣(2018年8期)2018-08-23
中小企业管理与科技·上旬刊(2018年12期)2018-02-18
科技创新与应用(2017年11期)2017-04-27
互联网天地(2015年1期)2015-12-04
科技资讯(2015年8期)2015-07-02
