
MXn半导体层状材料层间热电性质预测
2022-01-15 09:47:56陈敏敏崔丽玲陈灵娜
湖南工业大学学报 2022年1期
肖 金,陈敏敏,张 丹,崔丽玲,何 军,陈灵娜
(1.湖南工业大学 理学院,湖南 株洲 412007;2.南华大学 计算机学院,湖南 衡阳 421001)
1 半导体热电材料及其研究分析
把废弃的热能直接转换成电能,是缓解能源危机的一种绿色的、可持续发展的方案之一。热电材料利用Seebeck 效应能够实现热能与电能之间的转换。热电材料的转换效率通常用一个无量纲的ZT因子来衡量[1-4],ZT定义如下:

式中:S为Seebeck 系数;
σ为电导率;
κlatt是晶格热导率;
κe是载流子贡献的热导率;
T为温度。
载流子贡献的热导率可以被写成κe=LσT,其中L为洛伦茨系数,则公式(1)可以被写成
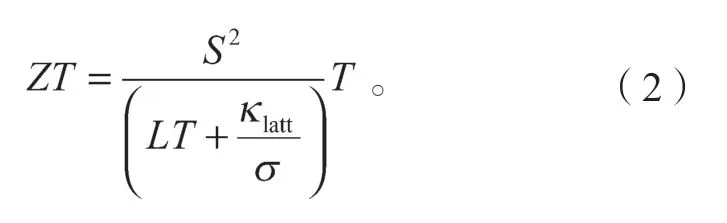
由式(2)所示ZT因子的定义公式可以看出,要提升热电材料的ZT,则其晶格热导率κlatt要越小越好,同时其电导率σ要越大越好。因此人们选择高性能热电材料的策略之一就是选择晶格热导率非常低的材料,然后在这些材料里选择电导率较高的材料。将当前常见的热电材料(如SnSe、PbSe 等[5-44])的ZT、晶格热导率和电子输运性质进行统计后,绘制成图1。图1中,圆圈的大小代表目前实验上测得的最高ZT值。名字前用#标示的代表该材料为层状材料。上标L、M和H均表示测量ZT和Klatt时的温度T,其中L表示T≤400 K,M表示400 K<T≤800 K,H表示T>800 K。//和分别代表层状材料中的面内方向和面外方向。图1中κlatt和ZT的数据参考来源见表1。图中MoS2和WSe2的晶格热导率数据为第一性原理计算数据。
表1 常见热电材料的κlatt 和ZT 实验数据参考来源Table 1 References of experimental data for κlatt and ZT of common thermoelectric materials
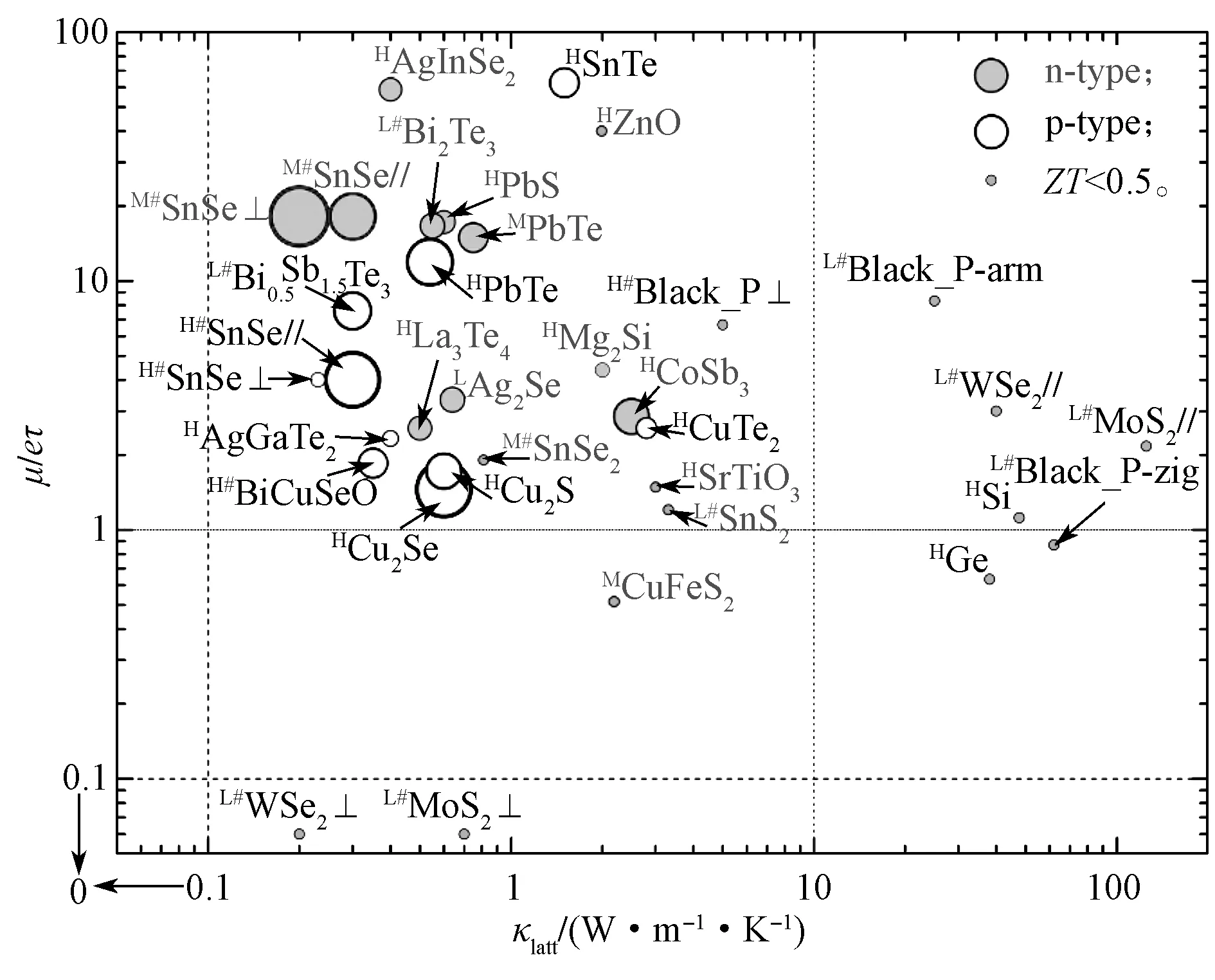
图1 部分块状热电材料的晶格热导率(κlatt)、电子输运性能(μ/eτ)和最优化的ZT 因子Fig.1 Lattice thermal conductivity (κlatt) and electron transport properties of bulk thermoelectric materials(μ/eτ) with an optimized ZT factor
通常材料的电子输运性能可以用有效质量m*来衡量(其单位为自由电子质量m0,1/m*=μ/eτ,其中μ为电子迁移率,e为电子的电量,τ为弛豫时间)。本研究中热电材料电子输运性能的实验数据参考来源见表2,其它材料的数据来源于TE DSIGN LAB[43-44]。
表2 热电材料电子输运性能的实验数据参考来源Table 2 References of experimental data for electronic transport performance of thermoelectric materials
从图1中可以发现,目前高ZT的热电材料的晶格热导率都低于10 W·m-1·K-1,同时电子的输运能力μ/eτ要大于1。这说明材料具有较低的晶格热导率和较高的载流子输运能力才能获得高ZT。正如前人提出的“声子玻璃,电子晶体”(phonon-glass electron-crystal)的概念[3],即声子传输象玻璃一样(玻璃对声子的传输非常差),而载流子的传输象晶体结构一样。
降低晶格热导率,可以通过削弱声子的传输或者增加声子的散射来实现。通常声子的传输与原子之间的相互作用相关联,如图2所示。而原子之间相互作用的强弱与原子之间的距离(d)成正相关性。有研究表明,通过极端高压可以使得MoS2层与层之间的间距减小,原子的距离接近会导致面外方向的晶格热导率明显增加[45]。对于共价键体系(原子之间的键长通常小于3 Å),原子的振动通过共价键在原子之间进行传递。在不考虑声子之间的散射时,其晶格热导率非常大。当原子之间的距离拉大后,原子与原子之间的相互作用会逐渐减弱,这会导致原子的振动在原子之间的传导变弱。因此,在一个体系中,如果存在近邻原子之间的相互作用并非化学键时,这个体系的声子传输将会被严重削弱[46],比如多重填充的方钴矿(multiple-filled skutterudites)[24]、BiCuSeO[20]等。这些材料在结构上可以被看成是一维链状或二维层状材料的聚合,链与链和层与层之间没有化学键相连接。实验上测得这些材料的晶格热导率都非常小(小于1 W·m-1·K-1)[20,23,46]。这说明大的原子距离可以导致很小的晶格热导率。
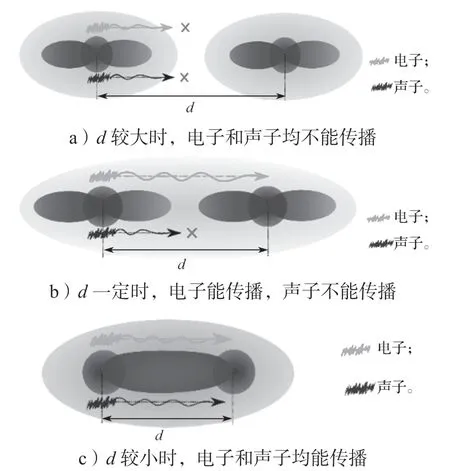
图2 声子和电子在不同原子间距之间的传输示意图Fig.2 Schematic diagram of phonons and electrons transports with various atomic distances
为获得高的ZT因子,除了低的晶格热导率外,还需要有较为合适的电导率σ。若电导率σ太大,则总的热导率主要由载流子贡献,比如金属材料。电导率σ太小,则材料几乎为绝缘体,也不利于热电之间的转换。在半导体材料中,由于禁带的存在,载流子的浓度可控且有限。所以半导体的电导率σ要比金属的低很多。同时Seebeck 系数通常在半导体材料中远大于金属材料。因此,高ZT因子的热电材料都为半导体材料。在半导体材料中,电子的传输与导带底波函数的延展性相关,如图2所示。波函数的延展性越好则有效质量越小,越有利于电子的传输。导带底通常是由原子轨道的反键态构成。原子之间反键态波函数的交叠也会随着原子距离的增大而削弱。在同一原子内,反键态的波函数空间分布范围通常要比成键态波函数的范围更宽。因此在某一个特定区间的原子距离范围内,原子之间的化学键已经断裂,但是反键态的波函数还是耦合在一起。通常,当原子之间的距离大于3 Å 时,可以认为化学键是断裂的。例如,在层状材料的黑磷中,其层与层之间的化学键是断裂的,但层与层之间的波函数仍然有交叠,这使得黑磷材料拥有非常好的层间载流子传输能力[47]。黑磷材料中磷原子之间的平均键长为2.26 Å,而层间磷原子的最小距离为3.59 Å。故可以认为声子的传输是处于“短程的”原子间距内(化学键长度范围内),而电子的传输则是“长程的”原子间距内(反键态波函数扩展范围内)。
在二维层状材料的每一层中,原子通过化学键结合在一起,而层与层之间则是通过层间的范德华(van der Waals,vdW)作用力聚合在一起[48-50]。范德华相互作用力与离子键和共价键不同,这种作用力很微弱,与距离相关,且易被扰动。声子在层状材料里沿面外方向(z方向)进行传输时非常困难。理论研究和实验测量都证明了这一点[31,51-52]。例如,在黑磷材料中,实验测得其室温下z方向的晶格热导率为(4.0±0.5)W·m-1·K-1,这比面内方向的晶格热导率至少要低一个数量级[31]。在WSe2中,其z方向的晶格热导率是面内方向晶格热导率的1/30[51]。此外,vdW 相互作用可以很方便地进行调控,例如建出vdW 异质节[53-56]和设计不同的层间堆栈结构[57-61]。实验上通过搭建无序的堆栈方式,使得WSe2面外方向的晶格热导率下降至0.05 W·m-1·K-1[52]。由于其优越的结构和独特的特性,vdW 耦合的层状材料引起了材料科研工作者们极大的关注[48-50,62-63]。在过去几十年里,大量的二维半导体材料被成功制备,例如二硫化过渡金属化合物(transition-metal dichalcogenides,TMDs)[64-65]、黑磷(black phosphorus,BP)[66-68]、第四族单硫化合 物(group-VI monochal-cogenides)[7,9,69-70]等。这些材料不仅在场效应晶体管、光电器件、电催化剂方面有着潜在的应用,而且在热电能转换方面拥有巨大的潜力[7,52-56,62-66,69-70]。
对于半导体层状材料,例如TMDs、BP 和SnSe等,目前的研究主要集中在这些材料的面内热电性质方面[7,31,71-78]。由于很多常见的层状材料中z方向的电荷输运能力与面内方向相比相差太远,因而存在一个误区,即认为z方向的载流子输运能力很差。这导致大家忽略了对层状材料中z方向性质的研究。比如,在MoS2材料中,其z方向的电阻是面内方向电阻的200 倍以上[79]。但是在黑磷材料中,沿z方向其能带结构有着非常大的色散关系[80],这使得其载流子的有效质量非常小[81-82]。黑磷中电子z方向的有效质量为0.12m0,空穴的有效质量为0.28m0[47]。通常有效质量越小,越有利于载流子的传输。在低温条件下,测得黑磷z方向的载流子迁移率可以达到8.5×103cm2·V-1·s-1[83]。最近,在n 型SnSe 材料中,因为三维的电子输运和二维的声子输运共同作用,使得z方向的ZT因子非常高,实验上测得其ZT值可以达到2.8±0.5(773 K)[8]。这些例子表明,半导体层状材料的z方向同样具有非常好的热电性质。但是在如此众多的半导体层状材料中,如何判断材料在z方向具有良好的热电性质呢?
本文将重点研究半导体层状材料的面外方向性质,回答什么样的层状材料具有电子可以传输但是声子的传输被禁止的特性。本课题组的理论研究结果表明,层状材料面外方向的晶格热导率都非常低。面外方向的电子输运可以分为如下两种类型:一种为赝层状材料,以SnSe 为代表,这类材料由于导带底反键波函数在层与层之间有明显的重叠现象,使得面外方向具有良好的电子传输能力;另一种为vdW 材料,由于其层与层之间没有反键波函数重叠,使得面外方向的σ非常低,如MoS2晶体。本课题组指出,在化学式MXn的半导体层状材料中(M 是阳离子,X 是阴离子),导带边缘通常由M 原子贡献。如果M 是过渡金属,由于导带底是非常局域的d轨道,不利于电子在z方向上的传输,例如TMDs。当M 是主要元素时,费米面周围的状态是pz轨道,这些轨道相对于d轨道来说要非局域一些,容易在层与层之间形成重叠。另外,波函数的交叠与原子之间的距离相关。相邻层之间M 原子的最小原子间距在脊状结构(如GeSe 和SnSe)中要比在三明治层状结构中小很多(如WSe2和MoS2)。因此,局域的d轨道和大的原子间距是vdW 层状材料在z方向的电子输运能力差的主要原因。而非局域的pz轨道和合适的原子间距,会使得赝层状材料在z方向有着较好的电子输运能力。低的热导率和高的电子输运能力使得赝层状材料在面外方向具有更好的热电性能。课题组的研究结果可以为在层状材料中选择出优异热电性能的材料提供指导。
2 研究方法
原子结构优化和电子性质的计算都是基于第一性原理的密度泛函理论。交换关联函数采用的是广义梯度近似下的PBE(Predew-Burke-Ernzerhof)方法。平面波的截断能设定为500 eV。计算过程中的收敛精度设定为所有原子受到的力小于0.01 eV/Å,同时能量的变化量小于10-5eV。计算过程中采用的是周期性边界条件。使用Monkhorst-Pach 网格方法对布里渊区进行取样,网格大小设定为11×11×3。对于范德华作用力的修正采用optB86b-vdW 方法[84]。通过结构优化后,GeSe 的晶格常量(a,b,c)为3.890,4.466,10.986 Å;SnSe 的晶格常量为4.163 ,4.475,11.587 Å。对于MoS2和WSe2,前者的晶格常量a=b= 3.152 Å,c= 12.360 Å;后者为a=b= 3.280 Å,c= 12.958 Å。所有计算得到的晶格常量与以前论文中报道的结果是一致的[85-87]。晶格热导率通过phono3py 程序[88]计算获得。计算中采用结构为2×2×2 的超胞。热电因子ZT的计算中,Seebeck 系数采用BoltZTraP 程序[89]计算获得。
3 计算结果与讨论
3.1 面外方向的晶格热导率
首先以SnSe、GeSe 和MoS2为例,计算层间的相互作用力与层间距的关系。通过计算不同层间距下体系的总能,把体系总能对层间距的一阶导数定义为层间的相互作用力(F=dE/dz)。作为参考,同时计算了氢分子体系中H 原子之间的作用力。所得结果如图3a 所示。
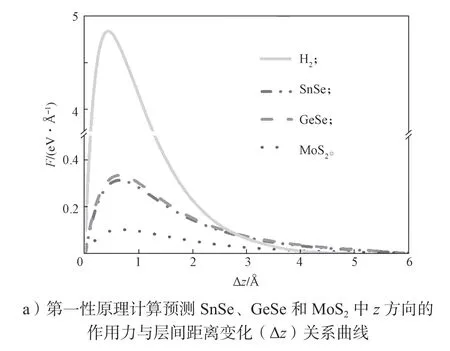
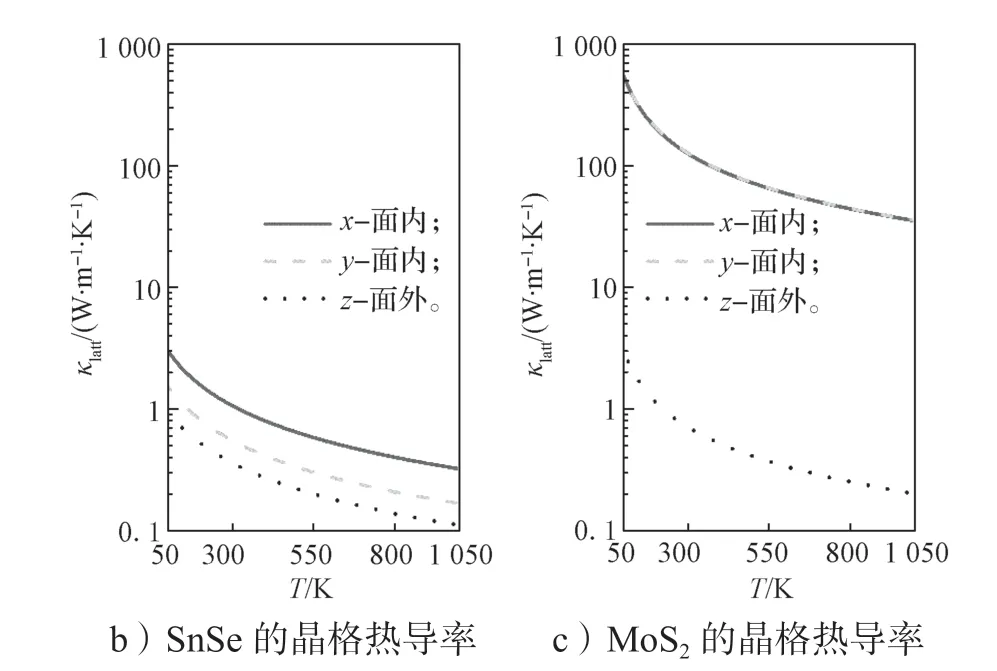
图3 层状材料中声子在面外方向(z 方向)的传输能力Fig.3 Transmission capability of phonons in the out-of-plane direction (z direction) in layered materials
从图3a 中可看出:1)随着层间距Δz的增大(对于H2则是原子距离),层间的相互作用力迅速衰减;2)层状材料中的层间相互作用力明显要比H2分子中H 原子之间的作用力微弱,因为前者是vdW 相互作用力,而后者是共价键。作用力的强弱决定着声子传输的能力。利用有限位移方法获得力学常数,通过基于线性声子玻尔兹曼输运方程的phono3py 程序包[88]获得材料的晶格热导率。SnSe 和MoS2的晶格热导率如图3b 所示。由图3b 可以看到,无论是SnSe 还是MoS2,面外方向(z方向)的晶格热导率是3 个方向中最小的。同时z方向的晶格热导率的绝对值都很低,在300 K 时,SnSe 和MoS2的晶格热导率分别为0.37,0.34 W·m-1·K-1。对于其它层状材料(如SnSe2、GeSe、SnS2等)都有相同的结论(部分层状材料在300 K 下的晶格热导率如表3所示),这主要是材料面外方向的相互作用力是范德华作用力,而面内方向则是由作用力更强的共价键彼此相连。
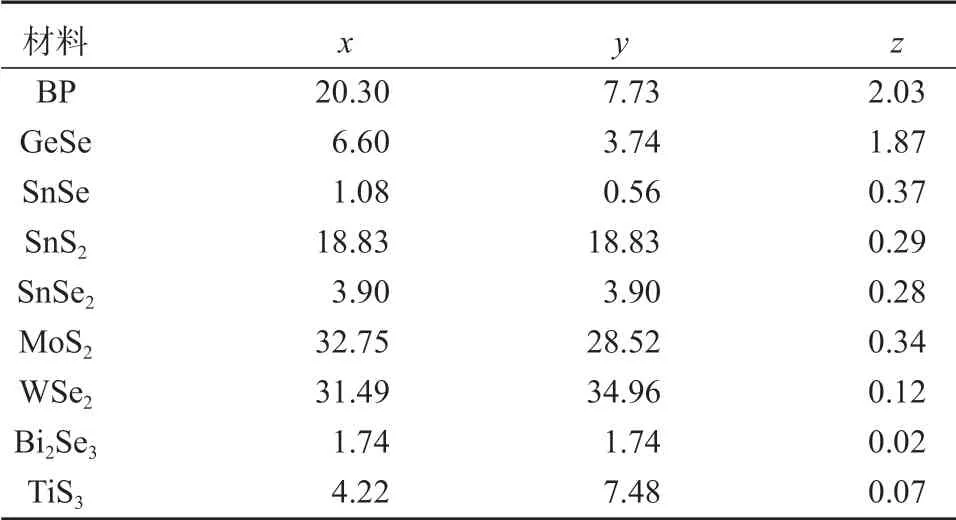
表3 部分层状材料在300 K 下的晶格热导率Table 3 Lattice thermal conductivity of layered materials at 300 K W·m-1·K-1
随着层间距的增大,面外方向的晶格热导率随之降低。例如,SnSe 中层间距增加0.25Å 和0.50 Å 时,300 K 下材料z方向的晶格热导率分别下降到0.19,0.11 W·m-1·K-1。上述计算结果充分说明,层状材料在面外方向非常符合“声子玻璃”的特征。那么在层状材料中能否找到“电子晶体”特征,这是确定高ZT热电材料的关键。
3.2 面外方向的电子迁移率
以SnSe、GeSe 和MoS2为例,图4给出了这3种材料z方向的电子迁移率随着层间距增大时的变化曲线。采用公式μ=eτ/m*计算材料z方向的电子迁移率,电子的有效质量m*是基于Γ-Z方向导带底的能带结构进行拟合获得的。这里将弛豫时间设定为τ=1×10-14s。从图4中可以看到,电子的载流子迁移率随Δz的增大而减小,这说明层间距越大越不利于电子在z方向的传输。另外,这3 种材料的载流子迁移率可以明显分为两种不同情况:一种具有非常大的电子迁移率,如SnSe 和GeSe;另一种的电子迁移率几乎为0,如MoS2。
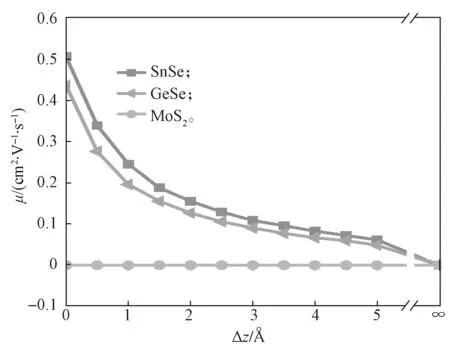
图4 层状材料面外方向的电子迁移率与层间距间的关系曲线Fig.4 Relation curves between out-of-plane electron mobility and layer spacing of layered materials
课题组进一步计算了其他层状材料面外方向的能带结构(图5)和电子有效质量(见表4)。
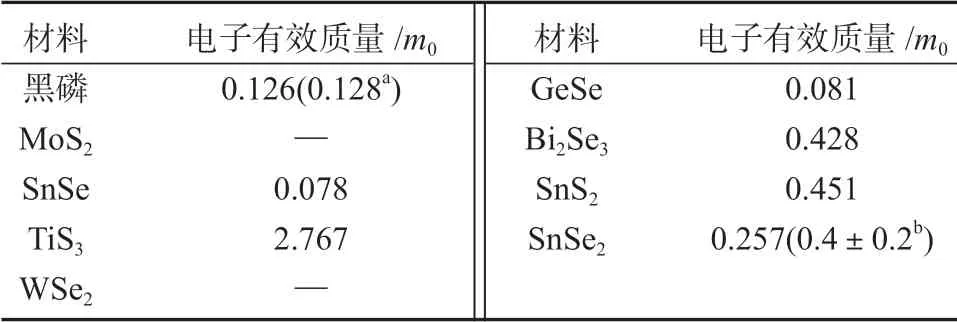
表4 部分层状材料中沿面外方向(Z-Γ)的电子有效质量Table 4 Effective mass (m*) of electron along Z-Γ of partial layered materials
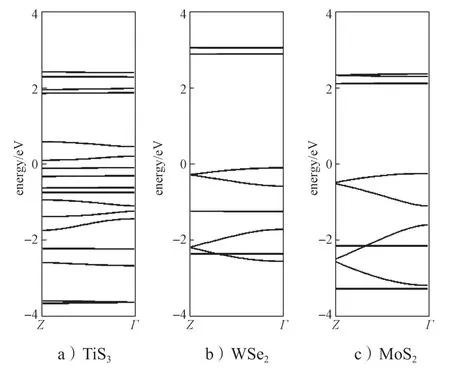
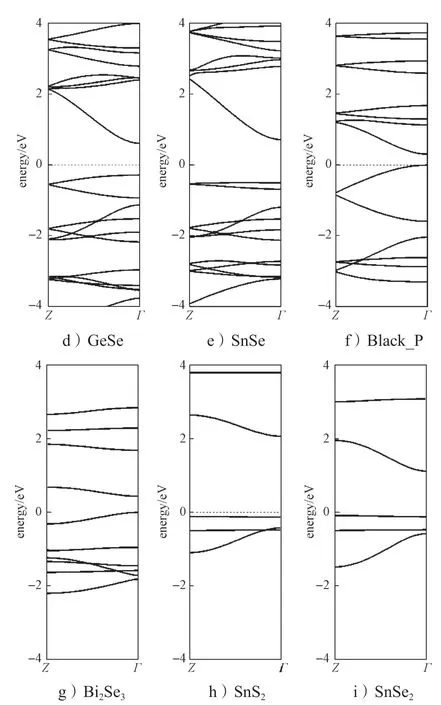
图5 部分层状材料面外方向(Z-Γ)的能带结构示意图Fig.5 Schematic diagram of band spectrum along Z-Γ of some layered materials
从图5所示层状态材料面外方向的能带结构图中可以看出,在Z-Γ方向(面外方向),TiS3、WSe2和MoS2材料的导带底基本上是水平能带,说明沿面外方向没有导带、没有色散关系,其导带底的波函数在层间是孤立的。水平的能带使得电子的有效质量非常大,电子在层间的传输只能通过跳跃进行,这极大地限制了电子在层间的传输能力。而SnSe 和GeSe材料在面外方向的导带有非常大的能量色散关系,这使得电子的有效质量非常小。大的色散关系表明导带底波函数在层间有交叠,其分布类似于晶体材料。因而在SnSe 和GeSe 材料中,电子在层间的传输具有晶体特征。
基于上述分析,可以根据电子在面外方向的传输性质好坏将层状材料分成两类:一类是具有良好层间电子输运的赝层状材料,如SnSe 和GeSe 等;另一类是电子在层间输运能量差的vdW 层状材料,如MoS2和WSe2等。
3.3 原子轨道耦合分析
接下来从原子轨道和原子结构方面分析为什么赝层状材料具有良好的z方向电子输运性质。图6所示为SnSe 和MoS2的投影能带结构和原子轨道耦合示意图。
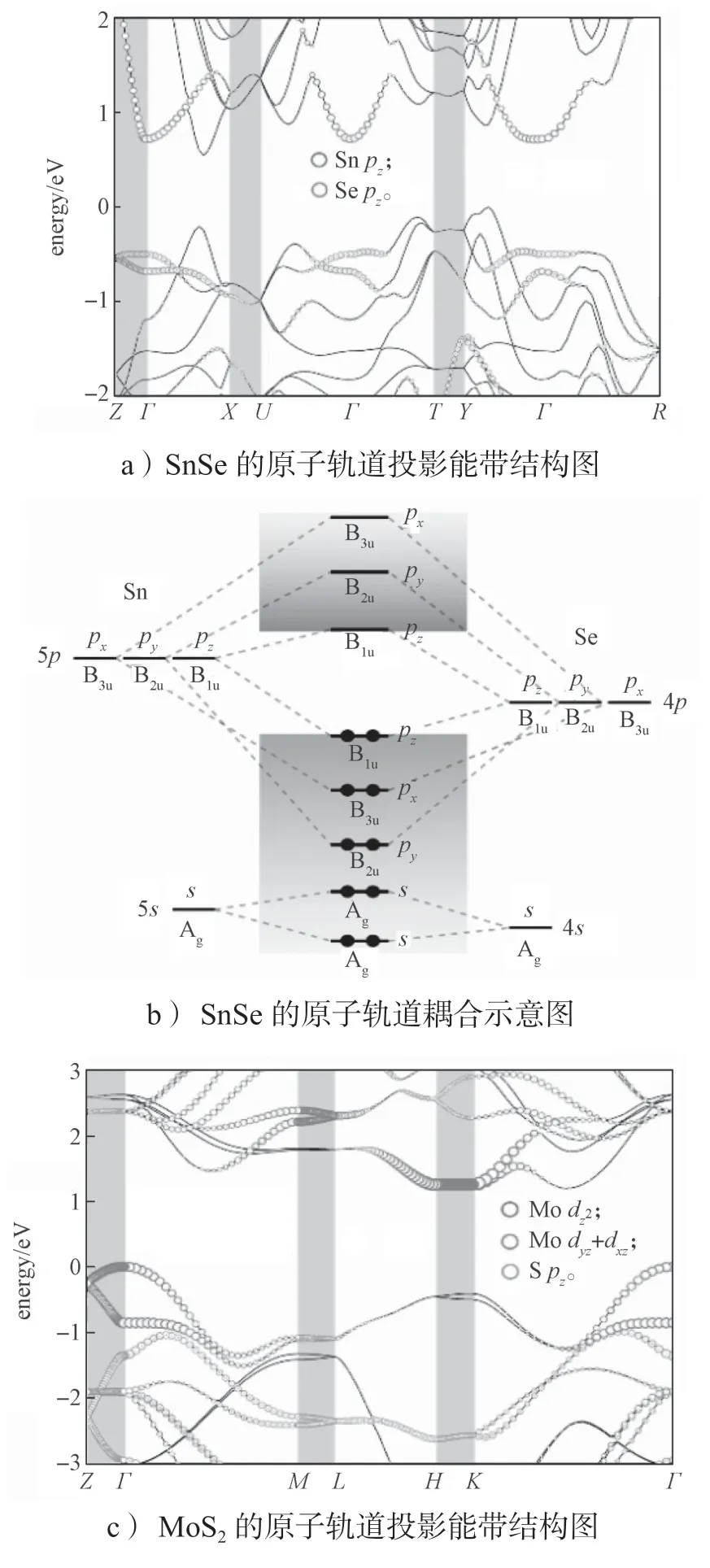
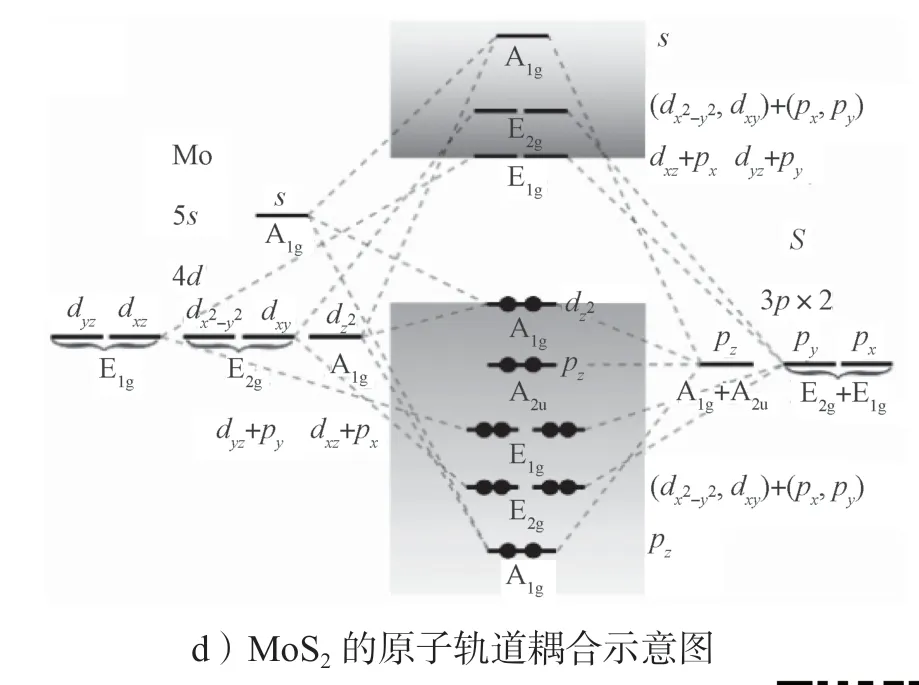
图6 SnSe 和MoS2 的投影能带结构和原子轨道耦合示意图Fig.6 Schematic diagram of projected band structures and orbital coupling of SnSe and MoS2

从图6a 和6c 所示能带带边的原子轨道贡献中可以看出,在SnSe 中Γ-Z的导带底(conduction band minimum,CBM)态是由Sn 原子的pz轨道组成,而在MoS2中,CBM 态主要是由Mo 原子的d轨道组成。图6b 和6d 是原子轨道耦合示意图。通过分析材料的对称性解释能带边缘(CBM 和VBM——valence band maximum)态的来源。对于层状材料,费米面附近的态会受到层间耦合的影响[91-92],但是所有的化学键都位于层内,层间没有化学键,因此原子之间的轨道耦合在块体结构和单层结构下是一致的。
对于SnSe 材料,块体SnSe 的一个原胞里包括了4 个Sn 原子和4 个Se 原子,其点群为D2h。单层的SnSe 的点群为C2V。SnSe 中阴离子(Se)数和阳离子(Sn) 数的比例为1:1。所以原子轨道耦合示意图中(图6b),只考虑一个Sn 原子的轨道与1 个Se 原子的轨道耦合。Sn 和Se 原子的价电子分别为5s25p2和4s24p2。在D2h点群下,s轨道的对称性表示为Ag,3 个p轨道的对称性分别表示为B3u、B2u和B1u。Sn 原子的5s轨道和Se 原子的4s轨道耦合,形成两个电子完全占据的Ag轨道,且远离费米能级。3 个p轨道中B1u轨道的耦合相对较弱,使得它的成键态为VBM,而反键态则为CBM。从VBM 往能量低处去,依次为px和py轨道。从CBM 往能量高处去,则依次为py和px轨道。在C2V点群下的情况和D2h点群下的情况完全一致。在SnSe 中,Sn 原子中的两个电子被Se 原子夺走,所以满带的轨道主要表现为Se 提供,而空带则主要是Sn 原子提供。
对于MoS2,这里只讨论Γ点的对称性。块体材料MoS2的原胞里包括2 个Mo 原子和4 个S 原子,其点群为D6h。单层MoS2的点群为D3h。MoS2中阳离子(Mo)数与阴离子(S)数的比例为1:2,所以原子轨道耦合示意图中为1 个Mo 原子与2 个S 原子耦合。Mo 原子和S 原子的价电子分别为4d55s1和3s23p4。在MoS2中S 原子的s轨道很深,基本上只在S 原子附近,没有参与轨道的耦合,所以在轨道示意图(图6d)中没有给出。在D6h点群下,Mo 原子的s轨道的对称性表示为A1g,原本5 重简并的d轨道劈裂成3 组:单重简并的A1g()、两重简并的E1g(dxz和dyz),以及两重简并的E2g(dxz和)。S 原子原本3 重简并的p轨道也分裂成两组,同时由于是两个S 原子,所以每组其对称性表示都有两个。S 原子pz的对称性表示为A1g+A2u,px和py轨道的对称性表示为E2g+E1g。Mo 原子的5s、轨道与其中一个S原子的pz有相同的对称性表示,这3 个轨道耦合使得轨道处于VBM。没有成键的A2u(另一个S 的pz轨道)紧随其后。E1g轨道耦合相对于E2g轨道耦合要弱一些。所以在A2u中,能量低一点的是E1g轨道(dxz与px耦合、dyz与py耦合)。比E1g轨道更低的则是E2g轨道(dxy和与px和py轨道耦合)。而对于空带,能量最低的是E1g轨道(dxz与px耦合、dyz与py轨道耦合),其次为E2g轨道(dxy和与px和py轨道的耦合)。所以CBM 为E1g轨道。对于单层的MoS2,其能带带边成分构成与块体结构一致,其轨道耦合也相同。其他高对称点下,MoS2中CBM 也都是由Mo 原子的d轨道构成。
由于CBM 态的构成成分不同,导致了SnSe 和MoS2在z方向的电子性质不同。在SnSe 中,CBM为Sn 原子的pz轨道,该轨道在空间上的分布成8 字形,且垂直于原子层面。这有利于相邻两层中的pz轨道形成交叠。在MoS2中,CBM 由d轨道构成,d轨道相对于p轨道来说,其电荷分布要局域很多。所以在相同的原子距离条件下,MoS2相比于SnSe来说难以形成层间的波函数交叠。从原子结构上看,SnSe 是一种脊状结构,Sn 原子位于每一个SnSe 子层的表面。相邻的SnSe 层中Sn 原子之间的最短距离(dM-M)为3.66 Å。这个数值比SnSe 中Sn—Se 键的平均键长2.78 Å 要稍微长一些。扩展的pz轨道加上较小的原子距离,使得波函数在SnSe 的层间有交叠。而在MoS2中,Γ-Z的CBM 态是由Mo 原子的dxz和dyz构成,H-K的CBM 态是 由轨道构成。虽然这些轨道的空间分布都有指向面外方向的分量,但是d轨道相对p轨道来说其局域性要强很多。同时,MoS2的原子结构是三明治结构,Mo 原子被上下两个S 原子层夹在中间。这种结构阻碍了Mo 原子d轨道在层间交叠。相邻层Mo 原子之间的最小距离dM-M达到了6.78 Å,约为 MoS2中Mo-S 平均键长(2.41 Å)的2.8 倍。局域的d轨道以及很大的空间距离,使得波函数不能够在MoS2中形成层间交叠。这符合图2中提出的物理模型。SnSe 有合适的dM-M,使得它在z方向能够传输电子。而MoS2有更大的dM-M,使其不能在z方向传输电子。如果把SnSe 的层间距拉大,则SnSe 在z方向的电子传输能力会随距离的增大而衰弱。通过计算发现,当Δz=3 Å 时,SnSe 中的dM-M数值和MoS2体系中的差不多,Γ-Z的CBM态也趋近于水平能带。经理论计算,得到Δz=3 Å 时电子载流子迁移率是Δz=0 Å 时的数据的1/5。可见,在Δz=3 Å 时的SnSe 已经和MoS2体系的性质接近。这种随Δz增大的变化趋势和图2中提出的物理模型非常吻合。
3.4 结果讨论
对于二元MX2赝层状材料,由于超低的晶格热导率和可观的电子迁移率,这类材料在z方向具有良好的热电性质。目前,大量的层状材料被发现或被制备出来[50]。可以将阳离子之间的原子距离和类型作为一个非常有用的划分这些层状材料的参数。MoS2是二硫化过渡金属化合物MX2(M 为Mo、W,X 为S、Se)中的一员。半导体TMDs 材料都具有相似的原子结构和电子结构[93]。在所有的半导体TMDs材料中,阳离子之间的dM-M都要大于6 Å。例如在WSe2中,W 原子之间的dM-M达到了7.80 Å。大的dM-M使得TMDs 材料在z方向的电子传输很弱。SnSe 和GeSe是第四族单硫化合物的MX(M 为Ge、Sn,X 为S、Se)中的一员[94]。这些材料的结构和电子性质都类似[95],它们的阳离子dM-M都在4 Å 以内,如GeSe和SnSe 的dM-M分别为3.46 Å 和3.66 Å,因此这类材料都可以划归于赝层状材料。最近的研究结果表明,人为增大SnSe 材料的层间距离会导致波函数层间交叠消失,使得电子层间传输性能急剧削弱[96]。随着材料中元素原子序数的增大,pz轨道会更加拓展,也越有利于波函数在层间形成交叠,从而导致电子在z方向的传输越顺畅。例如,SnSe 的z方向电子迁移率就高于GeSe 的。同时,原子序数增大使得原子质量更大,而大的原子质量会阻碍声子的传播,使得晶格的热导率更低[97-98]。课题组计算得到的300 K 下z方向的晶格热导率从SnSe、GeSe 到黑磷是依次升高的。SnSe、GeSe 和黑磷这3 种材料的原子结构相同,原子的质量是依次减轻的。因此,要选取好的热电性质的半导体层状材料,要求M 和X 越靠近元素周期表底部越好。通过第一性原理计算了几种层状材料面外方向随T变化的ZT因子,所得结果如图7所示。
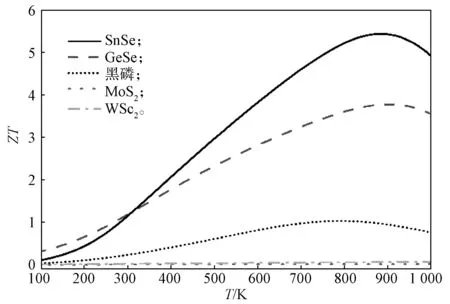
图7 几种层状材料面外方向的ZT 因子随T 的变化曲线Fig.7 Variation curves of ZT factor with T in out-of-plane direction of several layered materials
从图7中可以看出,同一温度下,SnSe 和GeSe材料面外方向的ZT值明显要高于黑磷、MoS2和WSe2的。这表明当材料原子结构相同时,元素质量越大,ZT越高(SnSe 大于GeSe)。
4 结语
本文提出了一个关于电子和声子的传输与原子距离间的关系模型。电子和声子的传输都随着原子间距的增大而衰弱,但声子传输衰弱得较快,而电子传输衰弱得相对要慢。在层状材料中,层与层之间是非常微弱的范德华作用力,这使得面外方向的晶格热导率非常低。两种非常典型的二元半导体层状材料(SnSe 和MoS2)的晶格热导率和电子结构计算结果表明,层状材料中面外方向(z方向)的晶格热导率非常小,是3 个方向中最低的。在SnSe 和MoS2中,声子在z方向的输运能力相当,但电子在z方向的输运能力存在较大差别,SnSe 的输运能力明显强于MoS2。在SnSe 中,CBM 的态由Sn 原子的pz轨道贡献,该轨道的电荷分布平行于z方向。同时,在SnSe 中,层间Sn 原子的最小距离小于4 Å。小的原子距离使得Sn 原子的pz轨道能够在层与层之间产生交叠,为电子在面外方向的传输提供通道。而在MoS2中,CBM 态是由局域的d轨道构成。同时Mo原子的层间距离很大(超过了6 Å),使得CBM 的态局域在层内。因此,可将CBM 态在层间有交叠的层状材料称之为赝层状材料。这类材料在声子传输上呈现二维特性,而在电子传输上表现为三维特性。因而,半导体赝层状材料,如SnSe、GeSe 等,在z方向具有超低的晶格热导率和可观的电子输运能力,是非常合适的高性能热电材料候选者。
猜你喜欢
储能科学与技术(2022年5期)2022-05-10 10:18:50
装备制造技术(2020年2期)2020-12-14 03:09:12
陶瓷学报(2020年5期)2020-11-09 09:23:04
数学物理学报(2019年5期)2019-11-29 07:46:50
重型机械(2019年3期)2019-08-27 00:58:44
数学物理学报(2017年5期)2017-11-23 07:51:09
潍坊学院学报(2016年6期)2016-04-18 13:56:55
焊接(2016年9期)2016-02-27 13:05:22
燕山大学学报(2015年4期)2015-12-25 02:19:40
新疆钢铁(2015年2期)2015-11-07 03:27:52
