
常染色体隐性多囊肾病合并青少年痛风一例并文献复习
2022-01-09 06:41梁锦坚李谦华邹瑶瑶郑东辉戴冽蔡小燕
岭南急诊医学杂志 2021年6期
梁锦坚 李谦华 邹瑶瑶 郑东辉 戴冽 蔡小燕
多囊肾病(polycystic kidney disease,PKD)是基因突变引起的遗传性肾病,以肾囊肿进行性增大、增多以及破坏肾脏正常结构为主要病理特征,是终末期肾病(end stage renal disease,ESRD)病因之一。PKD 分为常染色体显性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)及常染色体隐性多囊肾病(autosomal recessive polycystic kidney disease,ARPKD)[1]。
高尿酸血症(hyperuricemia,HUA)是痛风发生发展的基础。约2/3 尿酸经肾脏排泄,1/3 经肠道排泄,尿酸生成过多和(或)尿酸排泄减少可导致HUA。青少年原发性HUA 病因包括代谢综合征、遗传因素、生活习惯等;继发性HUA 的病因包括血液病或恶性肿瘤放化疗后导致尿酸生成过多,或慢性肾脏病、肾功能不全以及药物等因素尿酸排泄减少。
本文报道一例携带PKHD1 新突变的ARPKD合并痛风的青少年女性病例,提高对ARPKD 的认识,探讨特殊类型HUA 及痛风的诊断思路。
1 临床资料
1.1 一般资料先证者为2019 年9 月在中山大学 孙逸仙纪念医院风湿科住院的患者。家系成员包括先证者父亲及母亲。本研究为临床病例观察,经中山大学孙逸仙纪念医院伦理委员会批准(SYSEC⁃KY⁃KS⁃022),家系成员均知情同意并签署基因检测知情同意书。
1.2 研究方法(1)收集先证者及其父母临床表现、体格检查、实验室及相关辅助检查资料。对先证者进行长期随访,收集临床表现及辅助检查资料。(2)致病基因分析:采集先证者及其父母外周静脉血3 ml,经EDTA 抗凝后送迈基诺(MyGenos⁃tics)公司检测。采用二代测序(next generation se⁃quencing,NGS)对患者的标本全外显子基因检测,对NGS筛查的可疑基因突变位点,应用一代测序技术在患者及其父母样本中进行验证,对家系表型和基因型进行共分离研究。采用SIFT、PolyPhen⁃2、PROVEAN、MutationTaster 等生物信息学蛋白功能预测软件分别对新发现的突变进行功能预测,根据美国医学遗传学和基因组学学会(ACMG)指南对突变进行评级。(3)文献检索及复习:以“多囊肾病(polycystic kidney disease)”与“高尿酸血症(hyperuricemia)”为关键词组合在万方数据知识服务平台、CNKI 及PubMed 检索相关文献,检索日期为2010 年1 月1 日至2021 年7 月1 日,总结多囊肾病患者尿酸代谢情况及血尿酸水平对多囊肾病的肾功能影响。
1.3 统计学方法采用SPSS 25.0 统计软件进行数据分析,计数资料描述形式为例(%)。
2 结 果
2.1 临床资料先证者女,13 岁,因“发现海绵肾及血尿酸升高9 年,发作性关节痛1 次”于2019 年9 月入院。患者9 年前因“腹部包块”在中山大学孙逸仙纪念医院儿科住院,腹部超声及MR 均提示双肾海绵肾样改变。生化:血肌酐(SCr)72 μmol/L,血尿酸(SUA)441 μmol/L。尿常规:LEU 3+,WBC 106 个/μl。诊断为双侧多囊肾并尿路感染,抗感染治疗并复查尿常规好转后出院。2019 年7 月因右踝关节肿痛1 周、血尿酸715 μmol/L 到风湿科门诊就诊,查体:右侧踝关节肿胀,皮温稍高,伴压痛,局部无皮下结节。双肾区无叩痛。泌尿系超声示:双肾弥漫性肾损害改变,右肾窦分离扩张,右肾轻度积水。2019 年6 月起予别嘌醇0.1 qd 口服治疗,2 月后增加至0.25 g qd。为明确高尿酸血症原因收入院。查体:血压106/72 mmHg,身高153 cm,体重40 kg,BMI 17.08 kg/m2,发育正常,心肺腹及四肢关节查体未见异常。辅助检查:生化:SUA 561 μmol/L,SCr 89 μmol/L(1.006 mg/dL)。尿常规:比重1.012,pH 6.0,红细胞121 个/μl,白细胞15 个/μl。尿肌酐(UCr)6.4 mmol/L,尿尿酸(UUA)966 μmol/L。eGFR(Schwartz 公式)=0.55×身高(cm)/血肌酐(mg/dL)=0.55×153 cm/1.006 mg/dL=83.64 ml/min/1.73 m2。尿酸排泄分数(FEUA)=尿酸清除率/肌酐清除率×100%=(UUA/SUA)/(SCr/UCr)×100%=2.39%,FEUA<7%。既往史:否认高血压、糖尿病等慢性病史。月经史:12 岁月经来潮,平素月经规律。
2.2 家系调查先证者为独生女,父母非近亲结婚。父母无早发HUA、痛风病史,无多囊肾病史。
2.3 基因检测情况家属签署知情同意书,取患者及其父母静脉血送检,采用全外显子及Sanger测序验证进行相关基因分析。患者基因测序发现PKHD1 基因存在c.11314C>T 及c.4394T>G 突变两个杂合突变:(1)c.11314C>T(p.R3772X)为无义突变,在正常人群数据库中的频率为0.00060,为低频变异,HGMD 数据库已有该位点与多囊肾相关性的报道,ClinVar 数据库对该位点的常染色体隐性多囊肾病致病性判断为致病性/可能致病性变异。根据ACMG 指南,该变异判定为致病性变异(PVS1+PS1+PM2)。(2)c.4394T>G(p.V1465G)为错义突变,在正常人群数据库中的频率为0,为低频变异(PM2),HGMD 数据库未有该位点的相关性报道,ClinVar 数据库无该位点致病性分析结果;PolyPhen⁃2 分析示该突变可能有害(预测分数0.830,灵敏度0.74,特异度0.88),SIFT 分析示该突变有害(预测分数0.003,截断值0.05),PROVEAN分析示该突变有害(预测分数-4.99,截断值-2.5),MutationTaster 分析示该突变可能无害(PP2)。
经家系验证分析,先证者之母携带c.11314C>T(p.R3772X)位点变异(图1),先证者之父携带c.4394T>G(p.V1465G)位点变异(图2),家系基因检测结果证实突变和家系内患者共分离(PP1),c.4394T>G(p.V1465G)与前述致病突变c.11314C>T(p.R3772X)组成复合杂合(PM3),符合常染色体隐性遗传模式(PP4)。根据ACMG 指南,c.4394T>G(p.V1465G)判定为可能致病的变异(PM2+PM3+PP1+PP3+PP4)。

图1 先证者及其母亲PKHD1 基因存在c.11314C>T(p.R3772X)突变
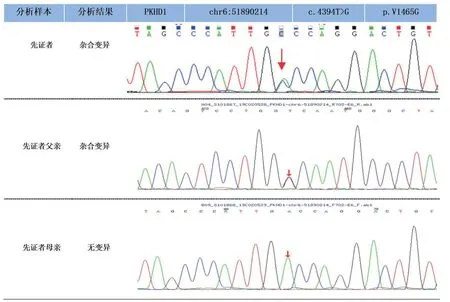
图2 先证者及其父亲PKHD1 基因存在c.4394T>G(p.V1465G)突变
2.4 随访完善基因测序后诊断为:(1)常染色体隐性多囊肾病;(2)继发性痛风性关节炎。继续予别嘌醇减少尿酸生成,别嘌醇剂量逐渐增加至0.5 g qd,随访1 年血尿酸可维持低于360 μmol/L,无关节肿痛发作,血肌酐、尿素氮及尿常规未见异常,监测血压未见异常。
2.5 文献复习检索到中文文献2 篇[2,3],英文文献4 篇[4⁃7]。除文献[4]外,其他文献PKD 病例的诊断依据为影像学及家族史,无基因检测结果,文献[3⁃7]纳入的PKD 均为ADPKD。PKD 患者HUA 发生率、血尿酸水平与肾功能水平的关系见表1。
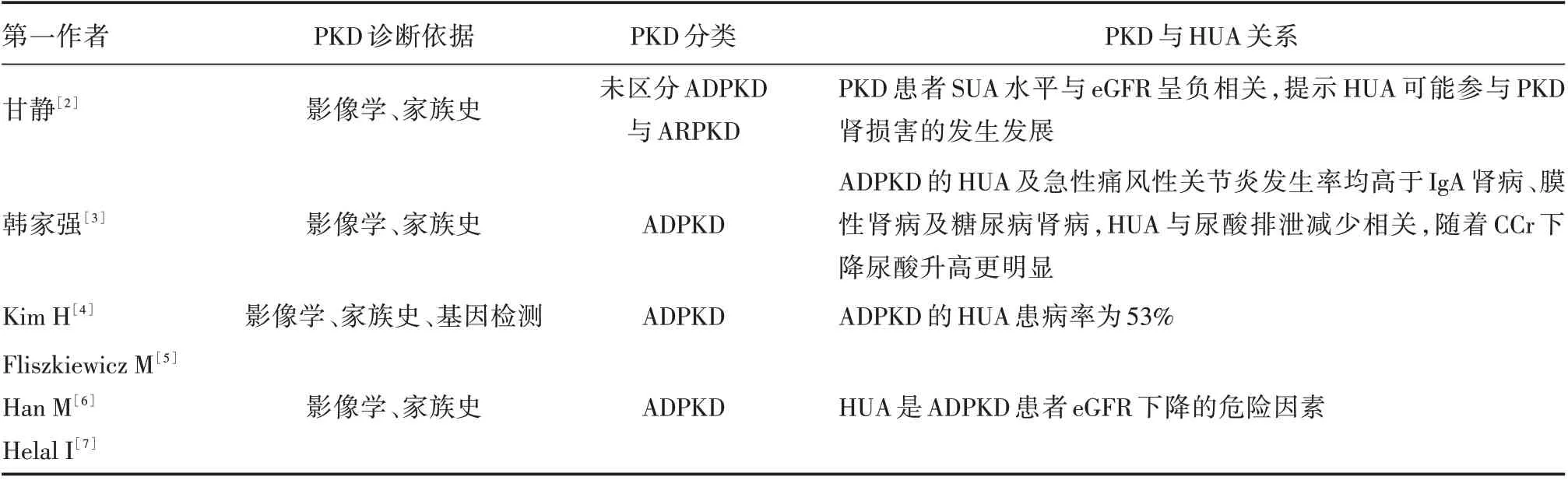
表1 PKD 与HUA 的文献复习
3 讨 论
ARPKD 又称婴儿型多囊肾,在胎儿期或新生儿期通过超声发现肾脏增大伴回声增强以及皮髓质界限不清。PKHD1 为ARPKD 的主要致病基因,其编码的纤维囊蛋白(Fribrocystin/Polyductin,PFC)主要在肾上皮细胞和胆管细胞中表达[1],FPC 缺陷可导致肾小管、胆管囊性扩张及纤维化。30%⁃40%的ARPKD 患儿在新生儿期因肺功能发育不全死亡,存活患儿可出现尿液浓缩功能减退、电解质紊乱、代谢性酸中毒及高血压等症状,肾功能随年龄增长进行性恶化,最终导致ESRD[1]。ARPKD 临床表现及病程进展差异较大,围产期、新生儿、婴儿、青少年及成人均可发病,围产期发病的患儿结局较差、死亡率高,新生儿期存活的患儿1 年及10 年生存率超过80%,大部分死亡病例发生在1 岁以内。部分PKHD1 基因变异类型与临床表型存在一定关联,双等位基因均为截短突变的患者肾脏受累更严重,存活患儿至少携带一个非截短突变[8]。本例患者儿童期超声已发现多囊肾病改变,经全外显子及Sanger 测序验证证实为PKHD1 基因突变所致ARPKD,突变类型为无义突变/错义突变杂合子,目前eGFR 正常,肾脏病变进展较缓慢,血压正常,暂未发现胆管扩张、肝硬化等其他相关病变。大部分ARPKD 基因型和临床表型难以关联,故目前仍不建议用具体的基因型来估测临床表型[1]。
过去原发性HUA及痛风以中老年男性多见,近年来由于生活水平提高及生活方式改变,HUA及痛风的发病呈年轻化的趋势。2015 年我国一项纳入15 省份成年居民的HUA 患病状况调查研究[9]表明其患病率为9.8%,男性血尿酸水平及HUA 发生率均显著高于女性,女性血尿酸水平及HUA患病率随年龄增长呈升高的趋势。雌激素通过调节肾脏转运蛋白促进尿酸清除,还可以通过低氧条件下抑制黄嘌呤氧化酶及维持脂质代谢稳定等途径减少尿酸生成[10],故女性在绝经期前发生HUA的比例低于男性。青少年HUA 发生率及性别分布情况与成人类似,于丽华等[11]对7~15岁青少年儿童进行调查发现HUA 发生率为10.7%,男性HUA 发生率显著高于女性。肥胖及不健康的饮食习惯是导致学龄儿童原发性高尿酸血症的原因之一[12]。本例患者为青少年女性,自幼发现血尿酸升高,无肥胖、血脂异常等代谢综合征的表现,全外显子测序未发现糖原累积症Ia、Lesch Nyhan 综合征、家族性青少年高尿酸性肾病(familial juvenile hyperuricemic nephropa⁃thy,FJHN)、髓质囊性肾病(medullary cystic kidney disease,MCKD)等与HUA相关的遗传性疾病相关的突变位点,但发现PKHD1 突变,结合其肾脏超声改变及家系资料,符合ARPKD诊断。
尿酸随血液进入肾小球时,几乎全部由肾小球滤过,大部分被近端肾小管重吸收,最后由远曲小管分泌而随尿排出。当肾脏存在病变时,尿酸经肾脏排泄相应减少。国内外尚无针对ARPKD 患者尿酸代谢及痛风发病情况的研究,大部分为研究纳入的病例为ADPKD,一方面可能与ADPKD 成年期发病多见及病程相对较长有关,另一方面可能与既往缺乏基因检测有关。本例患者eGFR正常,但FEUA提示其尿酸排泄减少,考虑与ARPKD 导致的肾小管功能损害所致。文献复习提示HUA 是PKD 患者eGFR 下降的危险因素,对于此类患者控制尿酸水平或有助于保护肾功能、延缓肾脏病变的进展。
综上所述,PKHD1 是ARPKD 的主要致病基因,突变位点及突变类型众多,亟待更多的研究来验证c.4394T>G(p.V1465G)突变的致病意义。此外,虽然HUA 及痛风发病有年轻化趋势,但临床上遇到非遗传及代谢因素相关的HUA 及痛风时,要注意排除继发性因素可能。
猜你喜欢
分子催化(2022年1期)2022-11-02
分析测试学报(2022年9期)2022-09-21
保健医苑(2022年1期)2022-08-30
中老年保健(2022年4期)2022-08-22
人生与伴侣·共同关注(2021年9期)2021-09-25
中老年保健(2021年8期)2021-08-24
华人时刊(2019年15期)2019-11-26
祝您健康(2019年8期)2019-08-09
生物学教学(2018年4期)2018-11-29
电脑知识与技术(2018年19期)2018-11-01
