
液液萃取-超高效液相色谱-串联质谱法定量测定污水中10种毒品
2021-12-31 03:05于丽丽储婷婷侯臣之王优美
中国药科大学学报 2021年6期
于丽丽,储婷婷,侯臣之,王优美,狄 斌**
(1中国药科大学药物分析系,南京210009;2国家禁毒委员会办公室-中国药科大学禁毒关键技术联合实验室,南京210009;3公安部禁毒情报技术中心,北京100741)
随着经济全球化和社会信息化加快发展,世界范围毒品问题泛滥蔓延,严重危害人类身心健康和家庭和睦,威胁社会治安,影响国家经济发展。《2019 世界毒品报告》显示,全球吸毒人数约2.7 亿,成瘾人数约3500 万,有约60 万人死于毒品滥用[1]。根据2019年中国毒品形势报告,截至2019年底,我国现有约214.8 万名吸毒人员,约占总人口的0.16%[2]。毒品滥用规模虽然整体呈现下降趋势,但目前依然较大,且越来越隐蔽的吸毒方式与吸毒活动私密性的加强使得排查难度加大。因此,掌握毒品滥用情况,全方位监控毒情态势,打击毒品犯罪,依旧是我国禁毒工作的重要内容。
在对不同来源的污水分析中发现,采用固相萃取进行前处理时,部分污水样本基质会严重影响待测物如吗啡、6-单乙酰吗啡(6-MAM)等在固相萃取小柱上的富集导致假阴性结果,或者有些成分在质谱检测时基质效应影响严重。因此,本研究建立了一种液液萃取-超高效液相色谱-串联质谱法(LLE-UPLC-MS/MS)同时检测污水中10 种毒品的定量分析方法。
根据我国毒品滥用情况,选择吗啡(MOR)、甲卡西酮(MC)、苯丙胺(AM)、甲基苯丙胺(MAM)、6-MAM、氯胺酮(KET)、3,4-亚甲基二氧基苯丙胺(MDA)、去甲氯胺酮(NK)、3,4-亚甲二氧基甲基苯丙胺(MDMA)、可待因(COD)作为毒品或其代谢物的毒情评估待测物。该方法操作简便、定量限低、重复性好,对于复杂基质样品同样适用,已较好地应用于国内10个城市的部分受基质影响的实际污水样品检测,期望成为常用的SPE-UPLC-MS/MS方法[3-12]的有力补充。
1 材 料
1.1 药品与试剂
10 种待测物及其氘代内标(包括MOR、MC、AM、MAM、6-MAM、MDMA、KET、NK、MDA、COD及MOR-D3、MC-D3、AM-D5、MAM-D5、6-MAM-D3、MDMA-D5、KET-D4、NK-D4、MDA-D5、COD-D6)储 备液均购于美国Cerilliant 公司。甲醇、乙腈(色谱纯,美国Tedia 公司);二氯甲烷(色谱纯,上海通用试剂公司);乙酸乙酯、氢氧化钠、甲酸铵、甲酸均为市售分析纯;超纯水由实验室超纯水系统制备。
1.2 仪 器
LC-30AD UPLC 超高效液相色谱串联LCMS-8050 质谱仪(日本Shimadzu 公司);Auto Science MTN-2800D 氮吹仪(天津Auto Science 仪器有限公司);THZ-312台式恒温振荡器(上海精宏实验设备有限公司);D3415R 低温冷冻离心机(瑞士BioTool公司);TGL-16M 台式高速冷冻离心机(上海湘仪离心机仪器有限公司);实验室超纯水系统(南京妙之仪电子科技有限公司)。
2 方 法
2.1 色谱条件
色谱柱 为Shimadzu Shim-pack C18(2.1 mm ×100 mm,1.9 μm);流动相:A为0.1%甲酸-5 mmol/L甲酸铵水溶液,B为乙腈;梯度洗脱程序:0 ~5 min,5% ~25% B,5 ~5.1 min,25% ~100% B,5.1 ~7 min,100% B,7 ~7.1 min,100% ~5%B,7.1 ~9.5 min,5% B;流速:0.4 mL/min;柱温:40 ℃;进样室温度:4 ℃;进样量:10 μL。
2.2 质谱条件
电喷雾离子源(ESI 源)正离子模式检测;雾化气(N2)流量3 L/min,加热气(N2)流量10 L/min,干燥气(N2)流量10 L/min,接口温度300 ℃,DL 温度250 ℃,加热块温度400 ℃;MRM 方式扫描;各待测物的MRM 离子对监测通道具体见表1。10种毒品及其内标的UPLC-MS/MS色谱图见图1所示。
2.3 样品液液萃取
取污水30 mL 至50 mL 离心管中,加入1 mol/L氢氧化钠溶液调节水样pH 至9.5 ~10.5,加入25 ng/mL 的工作内标溶液60 μL,充分振摇混合均匀。加入二氯甲烷-乙酸乙酯(1∶1)萃取剂10 mL,在恒温振荡器中以280 r/min,40 ℃振荡15 min,分取有机层,于50 ℃条件下氮吹至干,残留物用0.1% 甲酸水-乙腈(95∶5)溶液120 μL 涡旋复溶,经15000 r/min 4 ℃离心10 min,取上清液100 μL UPLC-MS/MS分析。
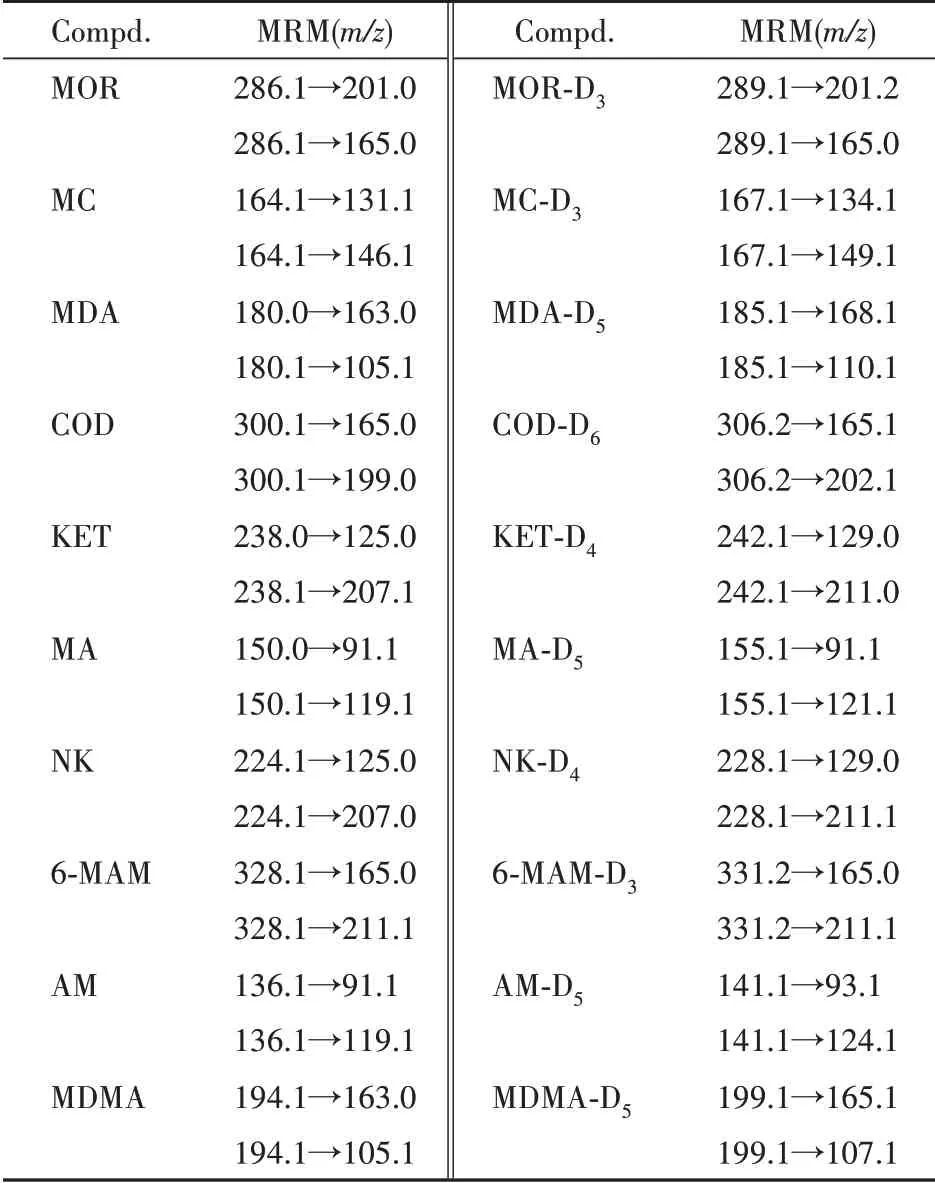
Table 1 MRM parameters of 10 illicit drugs and their internal standards
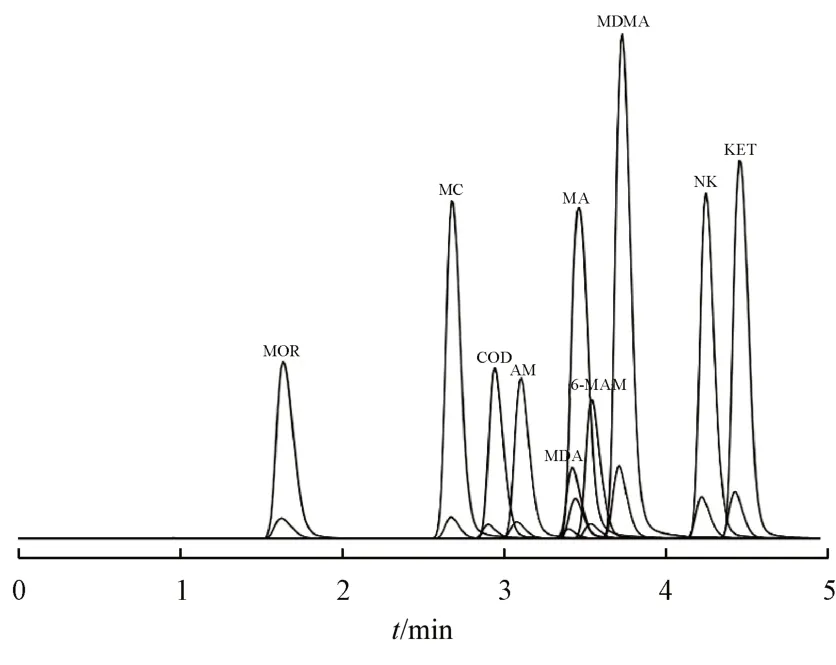
Figure 1 MRM chromatogram of 10 illicit drugs
2.4 方法验证
2.4.1 线性范围与定量下限 在混合空白污水中准确加入一定量的10 种待测物混合标准溶液,配制 成 质量 浓度 为1 ~500 ng/L(AM 为2.5 ~1250 ng/L)的系列样品,每个质量浓度双样本分析。按“2.3”项下操作后进样分析,以待测物的峰面积(Y)对待测物质量浓度(X,ng/L)进行线性回归运算(权重系数:1/X2),得到标准曲线方程。结果表明,10 种待测物在1 ~500 ng/L(AM 为2.5 ~1250 ng/L)范围内线性关系良好,其相关系数均≥0.9957。同时采用不含10 种待测物的混合空白污水样品配制定量下限(LLOQ)模拟样本,结果表明,当信噪比S/N 大于10 时,10种待测物的定量下限均为1 ng/L。
2.4.2 准确度与精密度 为了验证方法的准确度及精密度,向混合空白污水中加入一定量的10 种待测物混合标准溶液,配制成质量浓度分别为2.5、200、400 ng/L(AM 为6.25、500、1000 ng/L)的低、中、高质量浓度质控样品,每一质量浓度进行6 次重复实验,连续3 d 重复操作[12]。结果见表2,表明方法具有较好的准确度与精密度。
2.4.3 相对回收率 用混合空白污水基质配制2.5、200、400 ng/L(AM 为6.25、500、1000 ng/L)3 种质量浓度质控样品,每一浓度进行6 次重复实验,测得峰面积为A1;向复溶溶剂中加入相同量的混合标准溶液和工作内标溶液作为标准溶液对照组,每一浓度进行3 次重复实验,计算峰面积平均值为C。以峰面积A1 与C 之比计算各待测物的绝对回收率,同法计算各内标的绝对回收率,以待测物的绝对回收率除以内标的绝对回收率得到相对回收率,结果见表3。结果表明,各待测物经内标校正的相对回收率范围为96.36% ~106.43%,符合方法学验证要求。
2.4.4 基质效应 对混合空白污水基质30 mL进行液液萃取操作后,向复溶溶剂中加入同质控样品的混合标准溶液和工作内标溶液,每一浓度进行6 次重复实验,测得峰面积为A2。向复溶溶剂中加入相同量的混合标准溶液和工作内标溶液作为标准溶液对照组,每一浓度进行3 次重复实验,计算峰面积平均值为C。以每一浓度峰面积A2 与C 之比计算基质效应,同法计算各内标的基质效应,结果见表4。结果表明,各待测物基质效应符合方法学验证要求。
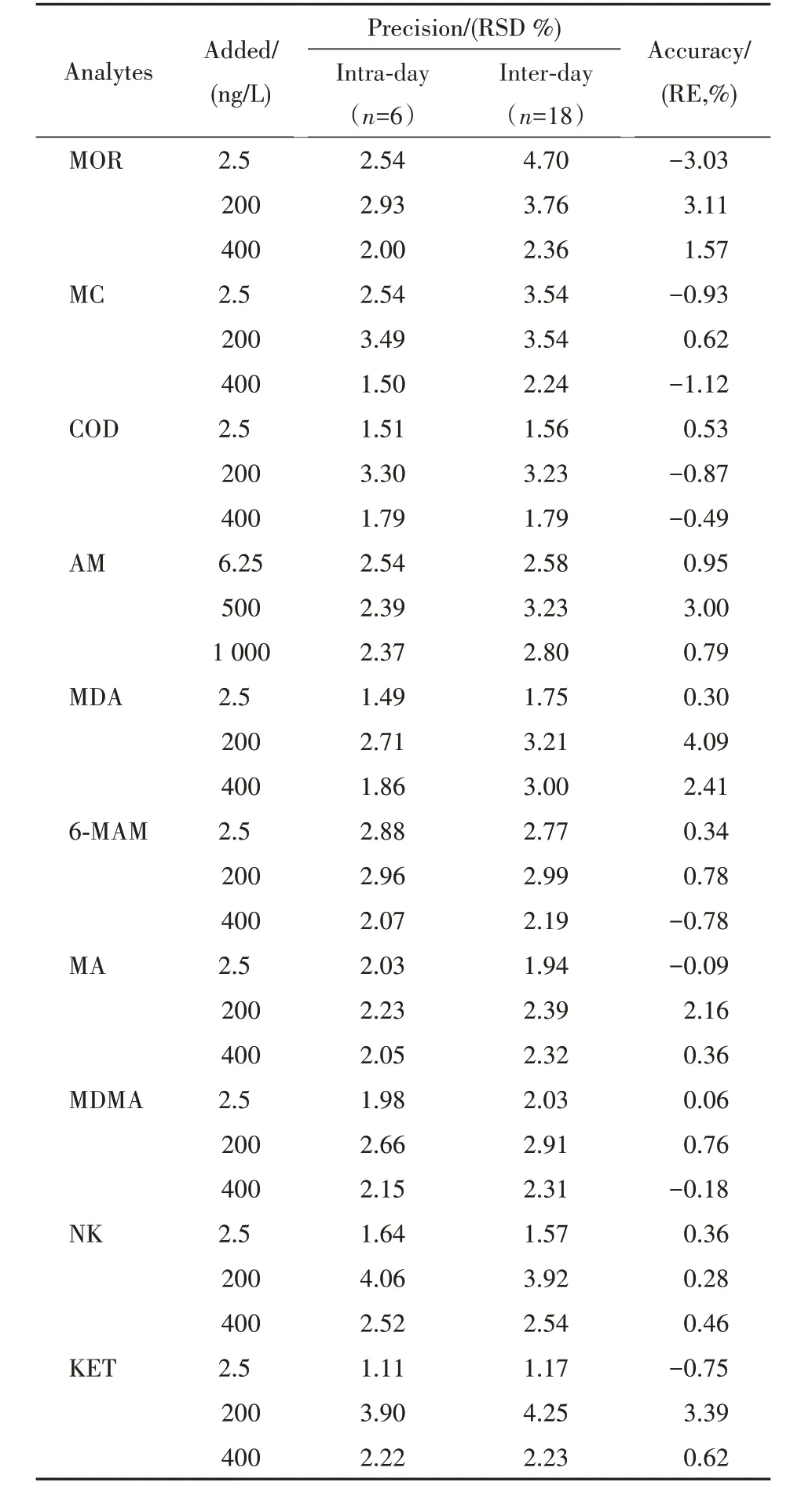
Table 2 Average precisions, accuracies at three concentrations of 10 illicit drugs (n=6)
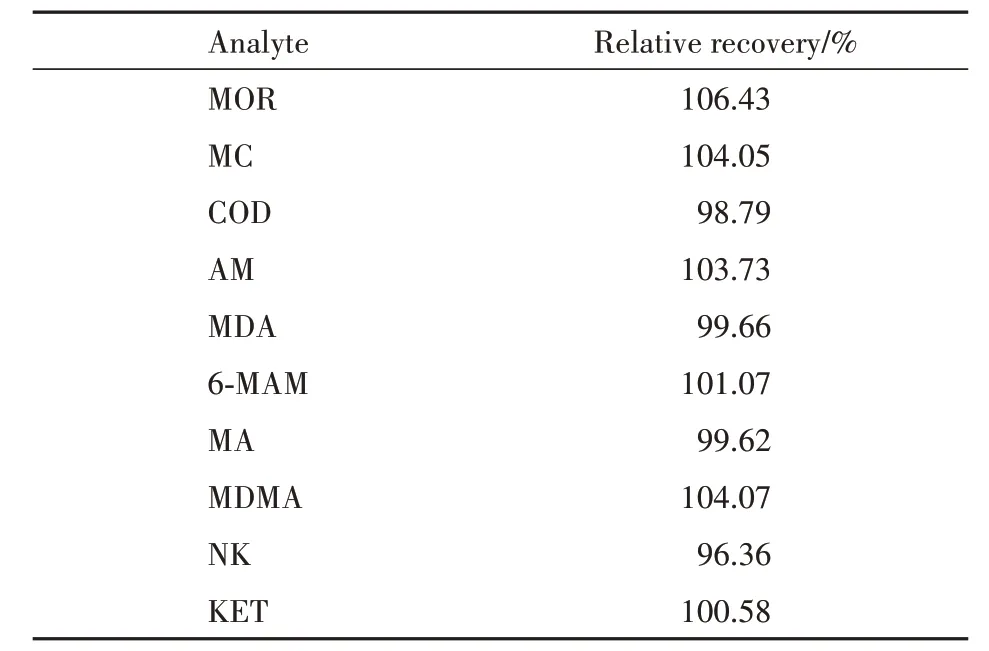
Table 3 Relative recoveries of 10 illicit drugs (n=6)
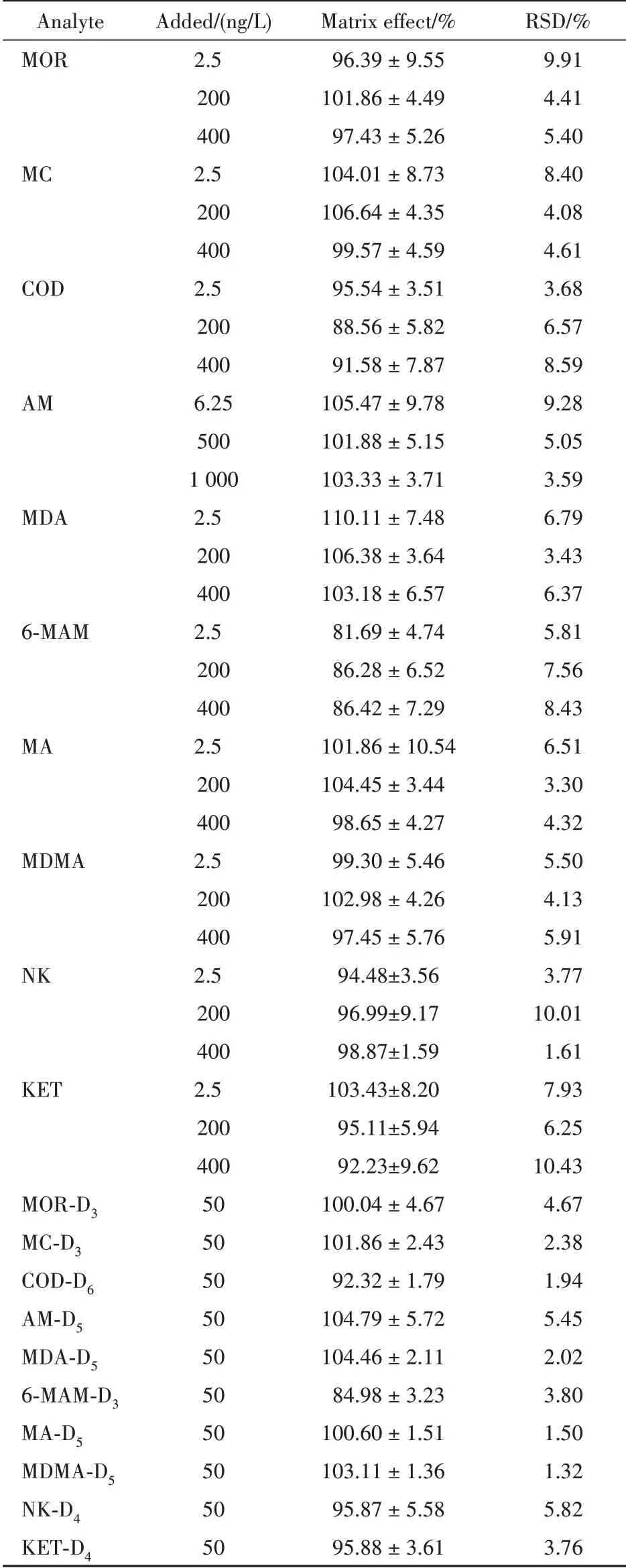
Table 4 Matrix effects at three concentrations of 10 illicit drugs and their internal standards (n=6)
2.4.5 进样稳定性 用混合空白污水基质配制低、中、高3 个质量浓度的质控样品,按“2.3”项下操作,分别在4 ℃进样盘放置24 h,每一质量浓度平行测定6个样本。结果表明,10种待测物样品在4 ℃条件下放置24 h稳定。
2.4.6 残留考察 取标准曲线最高点样品,将其与空白流动相样品交叉进样分析。结果表明,各待测物的残留响应值低于其定量下限的20%,并且不超过内标的5%,残留效应的影响可以忽略不计[12]。
2.5 方法应用
将本研究建立的LLE-UPLC-MS/MS 法应用于实际样品的检测,将采集自南京、江阴等10个城市不同污水处理厂的部分受基质影响的污水样品按“2.3”项操作后分析测定。这些样品应用SPEUPLC-MS/MS法[12]处理在萃取小柱上无法富集,待测物及内标都无响应,应用本研究建立的LLEUPLC-MS/MS 法进行测定,所有样品内标均可正常出峰,测得含量结果见表5。
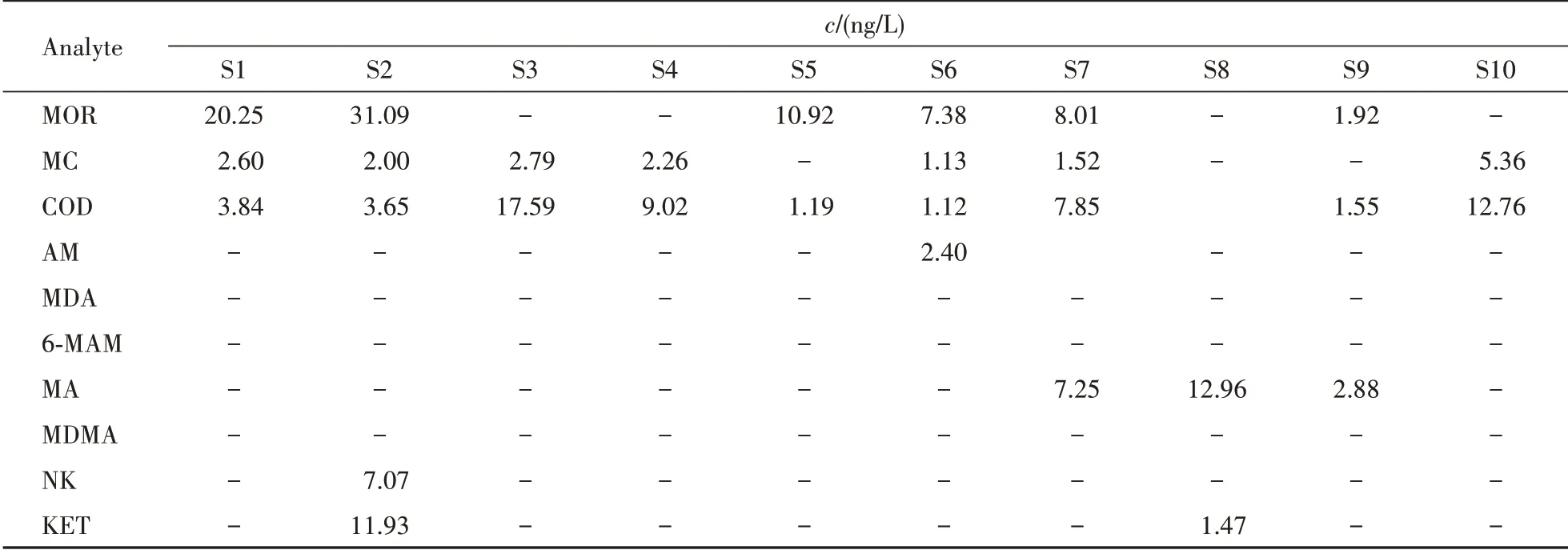
Table 5 Concentrations of 10 illicit drugs in wastewater samples (S1-S10)
3 讨论
3.1 色谱条件的优化
3.1.1 流动相的选择 采用0.1% 甲酸-5 mmol/L甲酸铵水溶液和0.1% 甲酸水溶液两种水相进行考察。实验结果表明,采用0.1% 甲酸-5 mmol/L甲酸铵水溶液作为水相时质谱响应提高,故选择0.1%甲酸-5 mmol/L甲酸铵水溶液作为水相。
3.1.2 流速的选择 对0.2,0.3,0.4 和0.5 mL/min 4种不同流速进行考察。实验结果表明,当流速为0.2 和0.3 mL/min 时,各待测物的峰形较宽且分析时间长;当流速为0.4 mL/min 时,峰形良好,且能在较短时间内满足分离要求;当流速为0.5 mL/min 时,部分待测物死时间出峰,故流速选用0.4 mL/min。
3.2 样品前处理方法的优化
3.2.1 萃取剂的选择 根据各待测物的性质及极性,分别对乙酸乙酯、正己烷、二氯甲烷、氯仿4 种不同极性有机溶剂单独及相互混合作为萃取剂进行考察,结果表明,二氯甲烷与乙酸乙酯混合萃取剂的萃取效率最高。接着考察二者不同混合比例,实验结果表明,二氯甲烷-乙酸乙酯(1∶1)为方法萃取剂时,各待测物总体萃取效果最好。
3.2.2 pH 条件的选择 根据各待测物的性质及pKa,分别对pH 8.5,9,10,11 这4 种不同污水pH条件进行考察,实验结果表明,在碱性条件下pH 10 时,各化合物总体响应最佳,故选择pH 10 为样品最佳萃取pH条件。
3.2.3 萃取剂体积的选择 采用这4种不同体积(5,10,15,20 mL)的萃取剂体积进行考察。实验结果表明,5 mL 萃取剂无法将待测物完全萃取出来;15 mL 萃取剂由于离心管中液体过满,振荡不充分,萃取效率不如10 mL 高;20 mL 萃取剂分成两等份,每次用10 mL 萃取剂萃取,合并两次萃取剂,所得峰面积与10 mL 萃取剂所得峰面积相近,考虑到减少有机溶剂污染,故选用10 mL为方法用萃取剂体积。
3.2.4 氮吹温度的选择 采用30,40,50,60 ℃这4 种不同氮吹温度进行考察。实验结果表明,30,40,50 ℃下氮吹后峰面积相近,60 ℃下氮吹后峰面积降低,考虑到节约时间,选择50 ℃为方法用氮吹温度。
4 总结
本研究建立了一种同时测定污水中10种毒品的液液萃取-超高效液相色谱-串联质谱定量分析方法。与常用的SPE-UPLC-MS/MS法[12]相比,该方法操作简单、定量限低、专属性好、重复性好,几乎不受污水基质效应影响,并已成功应用于10 个城市不同污水处理厂的部分受基质影响的污水样品检测,具有较高的准确度和可靠性。解决了实际测样中部分污水样品基质影响待测物在固相萃取小柱上富集的问题和质谱的基质效应问题。该方法可以成为SPE-UPLC-MS/MS 法的有力补充。这将为公安部门进行毒情监测提供重要的技术支持。
猜你喜欢
核化学与放射化学(2022年2期)2022-04-28
口腔护理用品工业(2021年4期)2021-11-02
饲料博览(2020年7期)2020-08-18
中国科技纵横(2019年23期)2019-02-14
海峡科技与产业(2017年1期)2017-03-04
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
中国信息化·学术版(2013年3期)2013-06-25
现代农业科技(2009年19期)2009-03-20
中学生数理化·八年级数学华师大版(2008年3期)2008-08-26
中国新闻周刊(2004年11期)2004-04-07
