
AVPR2 基因变异致先天性肾性尿崩症合并尿路扩张、生长激素缺乏1 例报告
2021-12-27 04:38刘晓静
临床儿科杂志 2021年12期
赵 玲 刘晓静
安徽医科大学第一附属医院(安徽合肥 230000)
肾源性尿崩症(nephrogenic diabetes insipidus,NDI)是由于先天性或获得性的病因,使远端肾单位中集合管细胞对精氨酸加压素(arginine vasopressin,AVP)受体不敏感,导致肾脏完全或部分失去尿液浓缩能力所致。先天性肾性尿崩症(congenital nephrogenic diabetes insipidus,CNDI)是由包括编码精氨酸加压素受体2(arginine vasopressin receptor 2,AVPR2)的X连锁基因变异,以及编码水通道蛋白2(aquaporin 2,AQP 2)的常染色体基因变异所致。约90%的患者是由AVPR2基因变异引起的X连锁隐性遗传性疾病,临床主要表现为多饮、多尿、持续性低比重尿,但也可同时合并尿路扩张和生长激素缺乏。本文报告1例经基因检测确诊的AVPR2基因变异致CNDI,并合并尿路扩张和生长激素缺乏患儿的临床资料。
1 临床资料
患儿,男,10 岁11 个月。因多饮、多尿伴生长迟缓6 年余收入院。患儿入院前6 年无明显诱因出现多饮、多尿,伴有烦渴,当时饮水量约3~4 L/d,尿量具体不详,夜尿1、2次,同时伴有身高增长缓慢,身高较同龄、同性别正常儿童低。随年龄增长,患儿饮水量及尿量渐增多,近2~3年饮水量约6~7 L/d,尿量与饮水量相当;身高明显低于同龄、同性别正常儿童。病程中,患儿饮食可,无头痛、视物模糊,无多汗、多食、体质量减轻,无发热及尿路刺激症状,大便正常。患儿系G3P2,足月顺产,否认出生时窒息或抢救史。父亲身高168 cm、母亲155 cm、哥哥172 cm;父母健康,否认近亲婚配,同胞哥哥身体健康,否认家族中有同类疾病史,否认家族性遗传病史,否认家族性肿瘤病 史。入院体格检查:体温36.6℃,脉搏96 次/min,呼吸24次/min,血压120/87 mmHg,体质量29 kg,身高128 cm(<P3),BMI 17.7 kg/m2,神清,精神可,正常面容,无明显脱水貌,皮肤黝黑,弹性可,心、肺、腹无异常,脊柱四肢无畸形,神经系统阴性。患儿10 岁9个月时于外院行精氨酸联合左旋多巴生长激素激发试验,示生长激素峰值<10 μg/L;同时查甲状腺功能五项提示正常;垂体MRI未见明显异常。入院后检查血常规、肝肾功能、血脂、胰岛素样生长因子-I、胰岛素样生长因子结合蛋白-3、皮质醇、促肾上腺皮质激素、胰岛素、肾素、血管紧张素、醛固酮、肿瘤标志物、尿红细胞形态、尿蛋白十一项均未见明显异常,25羟维生素D 11.1 ng/ mL,性激素六项示正常。禁水-加压素试验示禁水前血钠145.7 mmol/L,血渗透压307.72 mmol/L;禁水6小时后血钠149.3 mmol/L,血渗透压315.15 mmol/L。禁水过程中,尿比重持续低于1.005,血钠和血渗透压升高,尿崩症诊断明确。禁水6小时后皮下注射垂体后叶加压素3.0 U,2小时内留取3次尿液,尿量分别为750 mL、400 mL、300 mL,尿比重均<1.005,尿渗透分别为128 mOsm/(kg·H2O)、126 mOsm/(kg·H2O)、96 mOsm/(kg·H2O),确诊为肾性尿崩症。心电图示窦性心律。胸部正位片未见明显实质性病变。B 超检查示胆囊息肉;左、右肾集合系统分离,膀胱增大,膀胱壁毛糙;双肾积水,双侧输尿管扩张(图1)。静脉肾盂造影示双侧肾盂积水;左侧输尿管走行迂曲并扩张,右侧输尿管始终未见明确显影;膀胱充盈欠佳,未见明显充盈缺损(图2)。
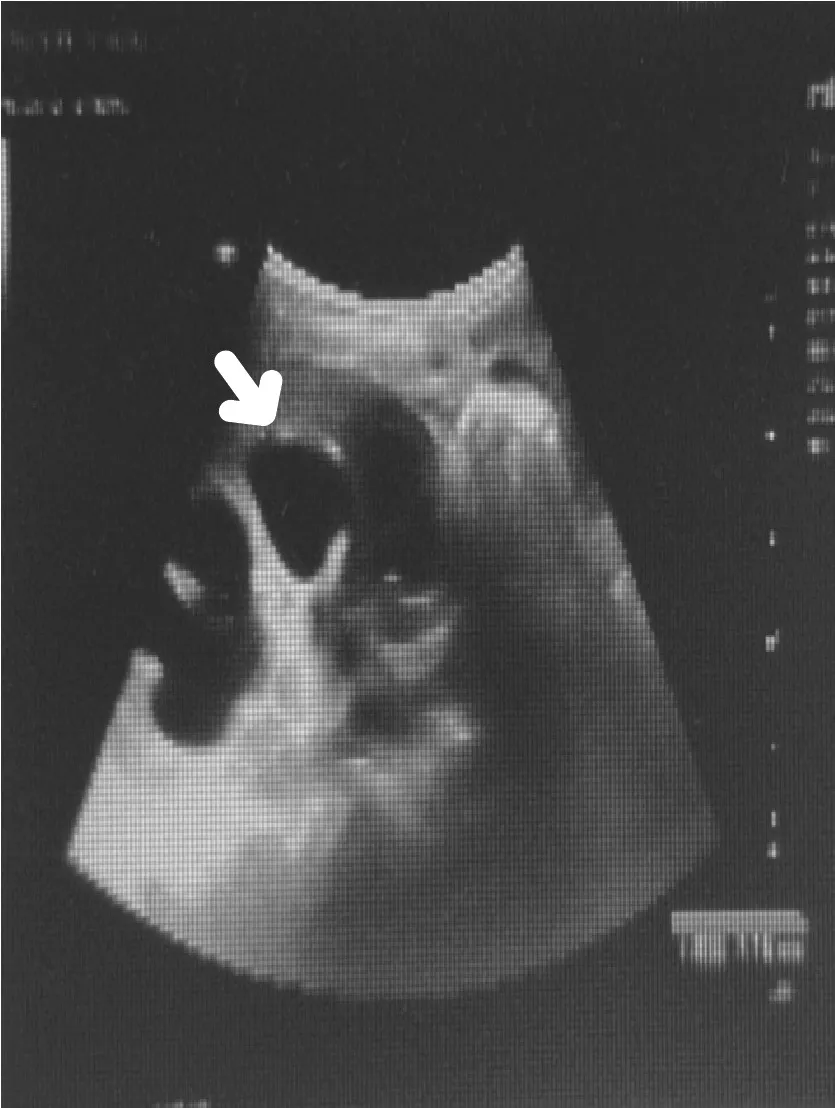
图1 患儿双肾、输尿管、膀胱超声检查结果
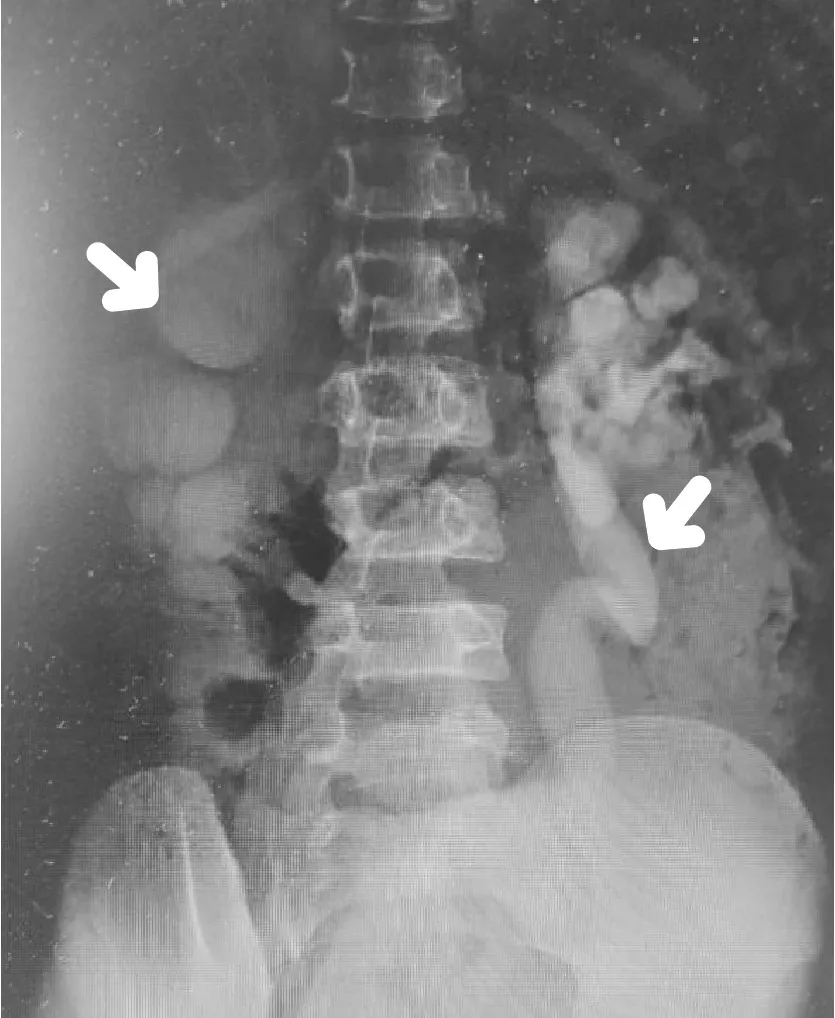
图2 患儿静脉肾盂造影结果
因患儿无继发性NDI的相关诱因,不能排除遗传性NDI,为进一步明确病因,获得家长知情同意,取先证者及其父母和哥哥外周血3~4 mL,送至北京智因东方转化医学研究中心北京全谱医学检验实验室行trio 全外显子组测序。结果显示,先证者AVPR 2基因第2 外显子区发生半合子变异c.347 A>T。该基因第347 位上的碱基A 变异为碱基T,导致氨基酸改变 p.K116 M(p.Lys116Met),即第116位上的赖氨酸变异为甲硫氨酸,为错义变异,系新发变异。根据美国医学遗传学与基因组学学会(ACMG)指南(2015年)[1],该变异为可能致病:PS2+PM1+PM2+PP3。其中强致病证据PS2为(家系)经双亲验证的新发变异;中等致病证据PM 1 为错义变异位于深入研究的无良性变异的外显子功能域,已确定的致病变异左右的错义变异且位于致病热点区;中等致病证据PM 2 为所有正常人群数据库频率<0.0005;可能致病证据PP3:两种预测软件(Provean、SIFT)预测出变异对基因(基因产物)有影响。家系验证分析显示先证者父母和哥哥均未携带该变异(图3)。
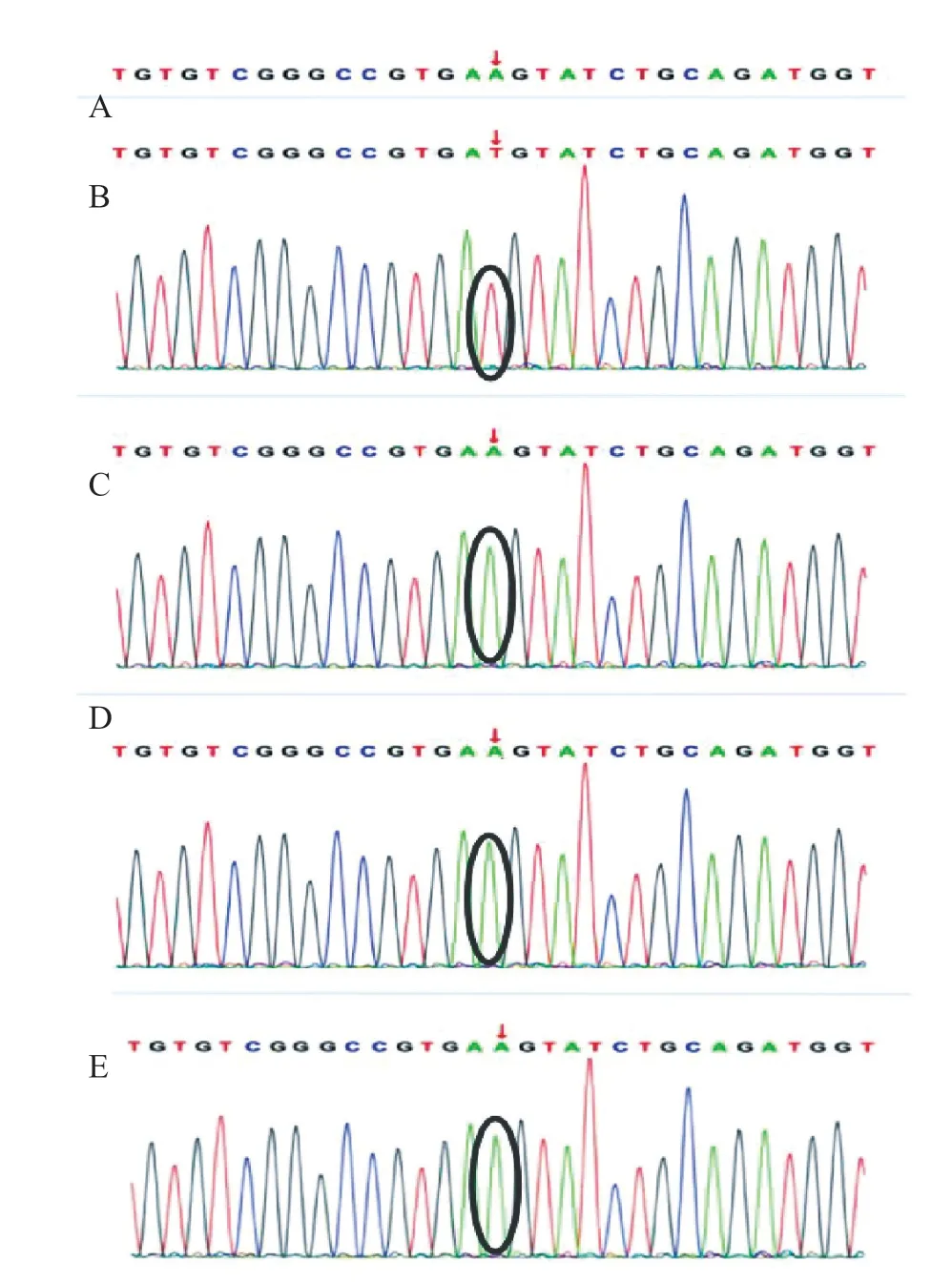
图3 先证者及其父母、哥哥基因测序结果
完善检查后予患儿口服氢氯噻嗪片、氯化钾颗粒、螺内酯片,出院时继续口服药物并保留导尿,定期监测血电解质及尿常规,必要时调整药物的服用剂量。2个月后电话随访,已拔除导尿管,出入量较前减少,约5 L/d。复查尿常规尿比重1.005,血钠139.2 mmol/L,血钾4.01 mmol/L;泌尿系超声示双侧肾盂、肾盏、输尿管扩张程度较前减小。待进一步控制尿崩症状,考虑予生长激素替代治疗。
2 讨论
CNDI 是一种罕见的遗传性疾病,多由肾小管远端集合管基底外侧膜表达的AVPR2基因变异引起,该基因位于染色体Xq28,编码由371个氨基酸组成的多肽,属于G蛋白偶联受体家族,是由7个跨膜区将4个细胞外和4个胞质区域相连而成[2-3]。
大多数AVPR 2变异导致AVP 受体(V 2 R)滞留于细胞内,影响AVP与受体蛋白结合,导致胞内信号转导异常,进而影响肾远曲小管的尿液浓缩功能。迄今为止,在AVPR 2基因中已经发现了250 多个可能的致病变异,包括错义变异、无义变异、小片段插入和缺失、大片段缺失和复杂重排,其中最常见的是错义变异[4]。由于AVPR 2基因变异具有异质性,CNDI的临床表现以及治疗反应也呈多样性[2]。婴幼儿期CNDI 起病隐匿,主要表现为发热、呕吐、烦躁、喂养困难等非特异性症状,随着年龄增长,出现多尿、多饮、烦渴、体质量及身高增长缓慢等典型症状,才会引起家长重视。以往报道还强调尿路扩张、行为异常等潜在并发症,如注意力缺陷障碍和严重的发育迟缓 等[5]。本例患儿即因AVPR2基因第2外显子的错义变异,导致自幼多饮、多尿,并伴生长发育迟缓,同时合并有尿路扩张。
CNDI患者并发尿路扩张和积水的病例在国内少有报道。曾有报道1例AVPR2基因变异合并梗阻性肾病的CNDI[6],1 例肾性尿崩症合并双肾积水,伴膀胱扩张[7],均因其他疾病就诊时意外发现双肾积水、输尿管扩张而确诊。日本一项有关CNDI的全国性调查中,173例NDI患者中有73例(42%)伴有泌尿系统并发症,并发现CNDI和获得性NDI在泌尿系统并发症发生率上无显著差异[8]。然而,CNDI更普遍伴有肾盂积水和/或输尿管积水。结合影像学检测、不同治疗后改善情况等的分析,目前一般认为尿崩症导致尿路扩张和积水的可能机制为长期多饮使肾脏产生的尿量不断增多,当在单位时间内超过输尿管排出量后,则会导致肾盂积水、输尿管代偿性扩张,加快尿液排泄进而缓冲肾盂压力。此外,膀胱高容量的排泄使膀胱逼尿肌代偿性肥大,导致输尿管远端壁内段相对狭窄而引起功能性梗阻,进一步使得上尿路失代偿性扩张。对于自幼发病者,可能与儿童膀胱三角区发育欠成熟及膀胱壁内输尿管移行部的长度较短有关,易使膀胱输尿管反流。最后,长期高容量的排泄使膀胱失代偿,尿液反流,不断加重肾盂和输尿管积水和扩张,同时肾实质因长期受到压迫而变薄,若治疗不及时可能导致肾功能严重损害[9-10]。
本例患儿的另一表现为生长迟缓,在CNDI 中并不少见。国内曾报道1 例合并生长激素缺乏的CNDI患者,未予生长激素替代治疗[11]。另有报道4 例肾性尿崩症中2例因AVPR2基因变异所致CNDI患儿存在身材矮小,经生长激素激发试验提示生长激素完全缺乏,予生长激素治疗,效果尚未报道[12]。国外曾报道1例CNDI男童,在6月龄确诊后予限钠治疗,期间出现生长不良;在患儿能够添加固体辅食后,适当增加饮食中钠盐量,实现了5.5 cm/年的生长速度,使患儿的身高和体质量保持在第25~50 百分位[13]。除了最初的喂养问题可能导致生长缓慢,摄入大量的液体也是原因之一,其会降低患儿食欲,引起慢性体质量下降,进而影响生长;此外,NDI 患者也有一些原因尚不清楚的并发症,如生长激素缺乏等。另一项对25例临床诊断为NDI患者的队列研究中,发现患者身材普遍矮小,但具体机制尚未明确[14]。生长激素的自然分泌量白天低于夜间,进入慢波睡眠后生长激素分泌陡增,并延续一段时间,入睡1 小时左右血中生长激素浓度达高峰,转入快波睡眠后又迅速减少,这种现象在青春期尤为显著。本例患儿身材矮小可能因夜尿影响其夜间生长激素的正常分泌,导致生长激素缺乏,引起生长缓慢。
CNDI 目前没有特异性治疗,对症支持治疗为首要治疗方式,如补充水分、保证低钠饮食、口服药物等,现常用的药物包括氢氯噻嗪、螺内酯等,用以减轻症状,缓解尿路扩张与积水,预防高钠血症、肾功能减退等并发症,进一步提高患者生活质量,延长寿命[15]。新的治疗方法,如利用分子伴侣进行的变异特异性治疗,已经在动物模型中进行了研究,但目前临床研究的数据很少,其目的是改善NDI的临床表型[16]。此外,基因靶向、变异特异性的治疗方法在CNDI 患儿的治疗中有巨大的潜能[17]。对于合并尿路扩张的治疗,主要包括药物控制尿量、短期导尿,多数患儿多尿症状改善后肾积水可减轻或消除;但对于严重的膀胱不可逆性扩大造成大量残余尿,同时继发输尿管梗阻,最终危害肾功能者,需进行手术治疗。无论何种方法,其治疗目标不仅为保护肾功能,而且要提高患儿的生活质量。其次,对于生长迟缓的儿童,经治疗后,大多数成年后仍难以发挥其全部的遗传潜能达到遗传身高,或达到人群的平均身高;而伴有生长激素缺乏者,虽然尚无相关机制报道,但生长激素治疗不失为一个重要选择,而开始使用生长激素的时机、停止治疗的标准及其收益风险比有待进一步研究[12]。
综上,CNDI在临床上除典型的多饮、多尿、烦渴等表现外,还可合并尿路扩张、生长迟缓等并发症。本例患儿多饮、多尿伴生长迟缓,影像学检查提示存在尿路扩张,基因检测结果示AVPR2基因c.347(exon2)A>T 变异,系新发变异,扩充了AVPR 2基因变异谱。但综合上述文献的结论以及本组病例,对于合并生长激素缺乏的患者,何时开始使用生长激素、停用标准以及效果仍有待探索,同时生长激素缺乏与CNDI 之间的关系也有待进一步研究。
猜你喜欢
现代临床医学(2022年2期)2022-04-19
河南外科学杂志(2021年2期)2021-12-09
临床超声医学杂志(2021年9期)2021-10-21
现代临床医学(2021年4期)2021-07-31
云南医药(2021年3期)2021-07-21
现代泌尿外科杂志(2021年2期)2021-03-02
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年6期)2020-02-01
医学研究杂志(2015年2期)2015-06-10
浙江中医杂志(2004年5期)2004-03-09
