
PAX2 基因变异致慢性肾脏病2 例报告并文献复习
2021-12-27 04:38孙晓朋聂娜娜张秋业王加兰尹讯章
临床儿科杂志 2021年12期
孙晓朋 林 毅 聂娜娜 张秋业 王加兰 尹讯章 常 红
青岛大学附属医院儿科(山东青岛 266000)
儿童慢性肾脏病(chronic kidney disease,CKD)是指各种原因引起的慢性持久性肾脏结构和/或功能损害,持续时间超过3 个月,部分患者可最终进展至终末期肾脏病(end stage renal disease,ESRD)。儿童CKD 发病率为(3.0~17.5)/1000000,患病率为(14.9~118.8)/1000000[1],而遗传因素是导致CKD 发生、发展的重要原因,儿童 CKD 中遗传性肾脏病占 42.1%~52.1%[2-3]。且儿童CKD 预后差,严重影响儿童生长发育及生存质量,是导致儿童ESRD的主要原因,已有研究表明,在儿童期接受肾脏替代治疗的患者几乎均诊断为遗传性肾脏病[4]。PAX 2基因是一种调控生长发育的基因,表达广泛,在泌尿生殖系统、眼、耳、中枢神经系统发育中发挥关键作用[5]。在人和小鼠模型中,PAX2基因变异可导致多种肾脏疾病,最常见的临床表型为肾脏发育不全[6],肾脏发育正常而单纯表现为CKD者少见。本文报告2例PAX2基因变异致CKD患儿的临床资料,并结合相关文献分析。
1 临床资料
例1,女,10 岁。于8 岁起病,因尿中多量泡沫就诊于当地医院,尿常规检查示尿蛋白阳性,后多次复查尿蛋白波动于+~++,遂来青岛大学附属医院(2018年)。患儿G1P1,足月顺产,出生体质量2.5 kg,无产伤或窒息史,生长发育及智力发育与同龄儿童相符。既往史无特殊,父母无血缘关系,父亲体健,母亲曾行尿常规示尿蛋白++,肾脏超声示双肾萎缩,肾发育不全。患儿入院体格检查:血压102/66 mmHg,体质量30 kg,无水肿,心、肺、腹查体未见明显异常。实验室检查:24小时尿蛋白排泄量14.3 mg/(kg·d);血尿酸643μmol/L,肌酐56μmol/L,尿素氮6.29 mmol/L;估算肾小球滤过率(estimated glomerular filtration rate,eGFR;根据改良的Schwartz公式)87 mL/(min·1.73 m2);血常规、血沉、抗链球菌溶血素O、抗中性粒细胞胞浆抗体、补体、抗核抗体、肝功能、电解质未见异常。泌尿系统超声示右肾窦内点状强回声,不除外尿酸盐沉积;未见“胡桃夹”现象。入院诊断为CKD(Ⅱ期),蛋白尿,高尿酸 血症。
入院后予患儿碳酸氢钠碱化尿液、肾炎康复片改善肾脏功能。患儿尿酸波动于422~519 μmol/L,尿蛋白波动于±~+。入院1个月时,加用苯溴马隆抑制尿酸在肾小管的重吸收、血管紧张素转化酶抑制剂(angiotensin converting enzyme inhibitors,ACEI)减少尿蛋白。患儿尿蛋白波动于-~±,尿酸269~334 μmol/L,肌酐60~70 μmol/L,尿素氮5~7 mmol/L。
因患儿持续蛋白尿、肾脏损害呈慢性过程,且有可疑家族史,考虑有遗传相关性肾损害可能。经患儿家长同意、医院伦理委员会批准,于2019 年8 月对患儿及其父母采血行DNA 全外显子二代测序,并对变异位点行一代测序验证。结果显示,患儿PAX 2基因存在杂合变异c.115C>T(p.Q39*),母亲基因分析结果与患儿相同,父亲PAX2基因无异常(图1)。该变异为无义变异,其编码区第115 位核苷酸胞嘧啶变为胸腺嘧啶,导致第39位密码子变为终止密码子,从而导致后续氨基酸缺失而不能合成完整蛋白质,致蛋白质功能受影响。该变异及致病性分析尚未见文献报道(参考数据库:HGMD Pro及PubMed)。根据美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)联合美国分子病理学会(American Society for Molecular Pathology,AMP)2015 年制订的基因序列变异的解释标准和指南进行致病性分析[7]:c.115 C>T 为无义变异,为致病变异(非常强致病性证据,PVS 1);比照千人基因组数据库(1000 Genomes)、人类基因变异数据库(HGMD)未见收录(中等致病性证据,PM 2);经多种算法预测会对基因或基因产物功能造成有害影响(支持致病证据,PP3)。综合上述c.115C>T变异的证据强度为“PVS1+PM2+PP3”,判断为致病性变异。该基因致病的遗传方式为常染色体显性遗传,患儿家系遗传方式符合遗传共分离。进一步对患儿外祖父母及舅舅行基因检测均未发现该基因变异,提示母亲为新发变异并遗传至患儿。
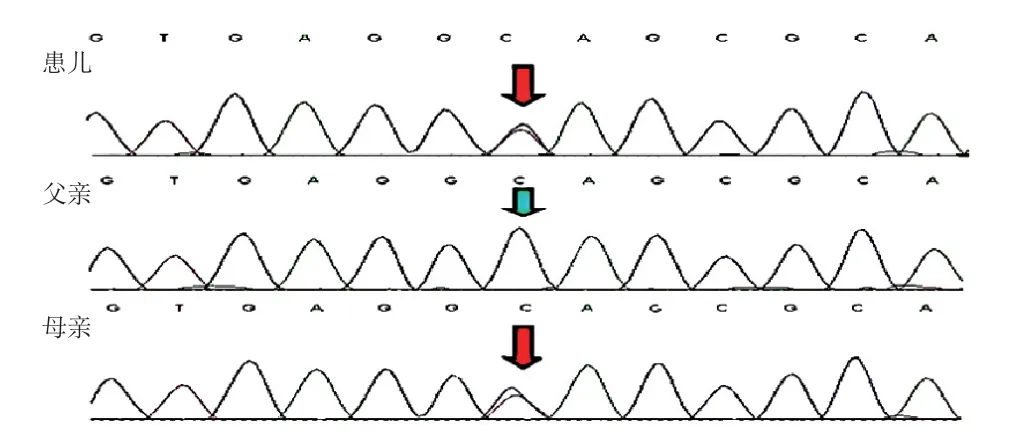
图1 例1 患儿及父母PAX2 基因 c.115C>T 变异测序图
例2,男,13 岁,G1P1,足月顺产,出生体质量 3.9 kg,无产伤或窒息史,生长发育及智力发育与同龄儿童相符。患儿父母体健,无血缘关系。患儿6 岁时(2014年)起病,表现为尿中泡沫增多。尿常规检查尿蛋白++,给予盐酸贝那普利减少尿蛋白治疗,并定期门诊随访,尿蛋白波动于+~++。患儿于2017年3月出现咳嗽症状,查血肌酐153.8 μmol/L,尿素氮 13 mmol/L,eGFR 34 mL/(min/1.73m2),遂收入院。入院体格检查:血压125/63 mmHg,全身无水肿,心、肺及腹部无明显异常,肾区无叩痛。泌尿系统超声示:双侧肾脏大小正常,未见明显畸形。肾穿刺活检示:肾小球肥大、系膜增生性肾小球肾炎,免疫荧光可见IgG,IgA免疫复合物沉积,见图2。入院诊断:CKD-Ⅲ期、系膜增生性肾小球肾炎。
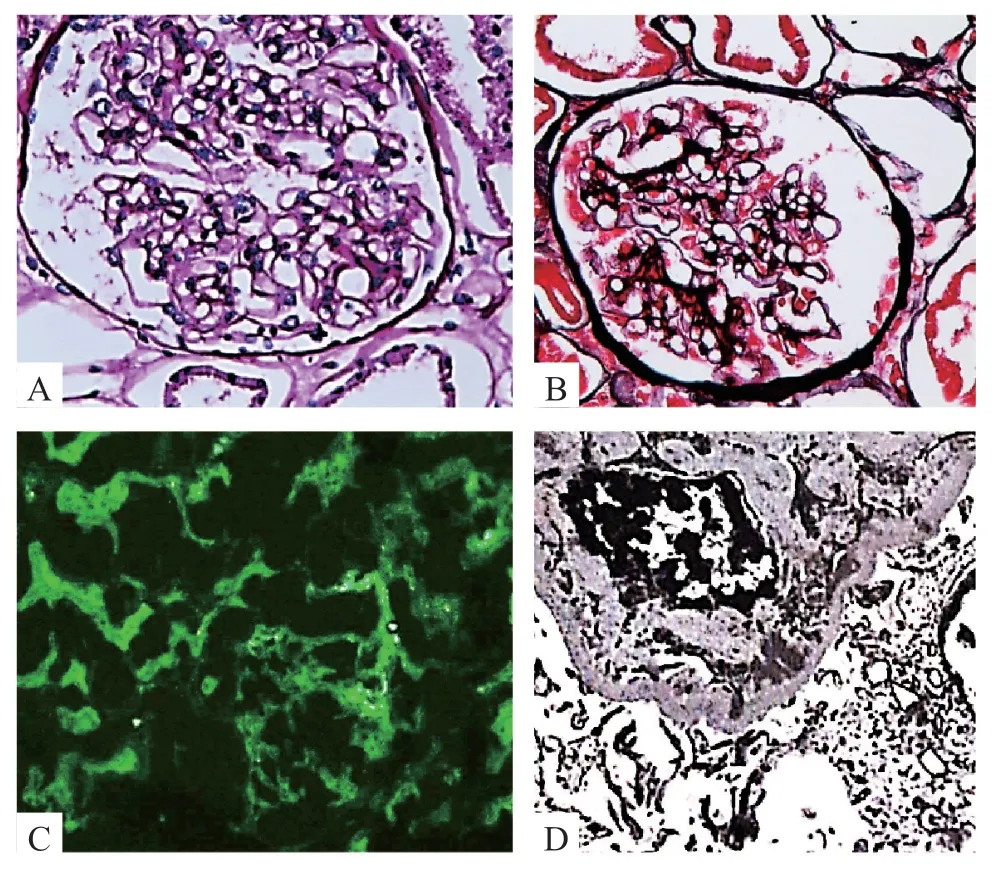
图2 例2 患儿肾组织病理
自2017 年4 月至2017 年10 月予患儿环磷酰胺(0.4 g×2 d)冲击治疗共7 个疗程,并予吗替麦考酚酯、甲基泼尼松龙治疗近1 年,口服ACEI。监测尿蛋白波动于±~+;肾功能逐渐恶化,末次复查血肌酐升至233.8 μmol/L,尿素氮升至14.6 mmol/L。期间曾有一次于腹泻后出现肾功能急剧恶化,后经抗感染、连续性肾脏替代治疗(continuous renal replacement therapy,CRRT)等治疗部分恢复。
考虑到患儿肾功能进行性下降,免疫抑制治疗效果不佳,为明确诊断,2018年8月行基因检测。结果发现,患儿PAX2基因存在c.986delC(p.T329Mfs*64)杂合变异(图3),其编码区986位核苷酸胞嘧啶缺失,导致第329个氨基酸由苏氨酸变成蛋氨酸,移码64个氨基酸后出现终止密码子,该移码变异会改变基因的开放阅读框,影响蛋白的正常翻译。该变异位点未见报道。根据ACMG 指南分析PAX 2基因c.986 delC变异致病性:c.986delC为移码变异,为致病变异(非常强致病性证据,PVS 1);比照千人基因组数据库(1000 Genomes)、人类基因变异数据库(HGMD)未见收录(中等致病性证据,PM2);经多种算法预测会对基因或基因产物功能造成有害影响(支持致病证据,PP3);经Sanger测序验证,该变异为新发变异(强致病性证据,PS 2)。综合上述c.986 delCT 变异的证据强度为“PVS 1+PM 2+PP 3+PS 2”,判断为致病性 变异。
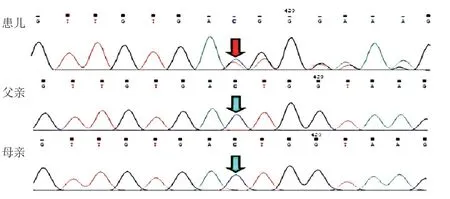
图3 例2 患儿及父母PAX2 基因c.986delC 变异测序峰图
2 讨论
以“PAX 2”和“chronic kidney disease”或“proteinuria”为关键词,对PubMed数据库,以“PAX2”和“慢性肾脏病”或“蛋白尿”,对中国期刊全文数据库、万方数据知识服务平台检索相关文献,检出临床资料和基因信息较全的文献9篇,其中中文文献1篇[8],英文文献8篇[9-16],包含32例患者,20个PAX2基因变异位点,其中12个错义变异,8个移码变异。32例患者均有肾功能异常、蛋白尿表现,16 例移码变异患者中有13例合并视神经异常、听力受损等肾外表现,而16例错义变异患者中仅7例合并肾外表现。患者起病年龄自出生前至成年期不等,除去11 例起病年龄不详者,余21例中19例起病年龄在20岁以内,部分PAX2基因变异患者产前就已出现肾脏发育异常,少数确诊时已经进展为ESRD,难以追溯既往是否有肾脏受累表现。14例患者行肾脏病理检查,其中5例为局灶节段性肾小球硬化(focalsegmentalglomerularsclerosis,FSGS),2例为系膜增生性肾小球肾炎(mesangial proliferative glomerulonephritis,MsPGN),其他还有肾小球肥大、肾小管萎缩伴间质纤维化、肾小球肾炎、非典型膜性肾病等。
在脊椎动物中,PAX 家族共有9 个成员,其中PAX2蛋白含有396 个氨基酸,由PAX2基因编码,是一种调控生长发育的关键转录因子,参与多个胚胎器官的发育过程。人类的PAX 2基因位于10 号染色体长臂24~25区,由11个外显子组成,跨越约70 kb[17]。PAX 2基因变异所致临床表型复杂,除肾脏发育不全及眼部异常外,还可出现内耳和神经系统受累,如高频听力损失、癫痫发作等[18]。PAX 2基因变异最常见临床表型为肾-视神经盘缺损综合征(renal coloboma syndrome,RCS),即以先天性肾脏和泌尿道发育异常、视神经盘发育不良为主要临床表现。汇总现有文献,肾脏发育异常、眼睛受累及高频听力损失所占比例分别为92%、77%和7%[19]。近几年,髋关节发育不良、先天性心脏缺陷、骨骼异常亦有报道[8,15]。本组2例患儿及例1 的母亲均仅有肾脏异常表现,而无任何视觉及听觉损害,不符合RCS诊断。PAX2基因变异患者表型中非RCS类型相对少见,但在孤立性肾脏异常如肾发育不全或FSGS 患者中发现PAX 2基因变异逐渐增加[18]。有报道1例10号染色体10q23.2q24.3区域缺失导致PAX2基因完全缺失患者,为人类第1例PAX2基因杂合子缺失伴肾脏异常,而非RCS病例[21]。
本文例1患儿PAX2基因变异遗传自母亲,两者基因型相同,表型亦相似。患儿母亲具体发病年龄不详,目前肾脏萎缩且持续存在尿蛋白,因例1 患儿无明显肾脏发育畸形,考虑母亲肾脏萎缩可能与长时间蛋白尿所致肾功能损害有关。分析携带PAX 2基因变异家系的临床表型发现,尽管有相同的基因型,患者的起病年龄、累及脏器、病变严重程度上有较大差异,单倍体剂量不足和表观遗传因素可能是临床表型差异的原因[10-11]。
在肾脏受累方面,目前已报道的肾脏病理改变包括FSGS、MsPGN等。PAX2基因变异的成年患者中有60%肾脏病理表型为FSGS[12]。Wilm肿瘤蛋白l(Wilms’ tumor 1,WTl)作为一种转录调节因子,在肾小球足细胞内持续表达,对维持正常的足细胞功能起着重要作用。研究发现,WT1的正常表达受到PAX2基因的调控,PAX 2变异将下调WT 1 的表达从而导致FSGS 的发 生[22-23]。本文例2患儿肾脏病理表现为MsPGN,该表现较为少见。研究显示,肾脏病理类型如MsPGN、非典型膜性肾病等,可能是由于患者年龄小、尚处于疾病早期所致,亦或PAX 2基因变异本身引起多种肾脏病理表型[15]。
PAX2基因变异所致肾脏疾病并无特殊治疗方法,本文例2 患儿应用激素及免疫抑制剂治疗后,肾功能未见明显好转。本文2例患儿经ACEI类药物治疗后,尿蛋白较前减少,肾功能相对稳定。研究报道,ACEI及血管紧张素Ⅱ受体阻滞剂类药物治疗后,有助于减缓PAX2基因变异所致儿童期CKD进展[10]。
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
中国典型病例大全(2022年11期)2022-05-13
中国土壤与肥料(2021年5期)2021-12-02
天津医科大学学报(2021年4期)2021-08-21
家庭医学·下半月(2018年9期)2018-12-10
老友(2017年12期)2018-01-23
中国动物保健(2015年4期)2015-10-21
医学研究杂志(2015年9期)2015-07-01
老友(2010年9期)2010-09-16
浙江中医杂志(2004年3期)2004-11-20
