
儿童原发性远端肾小管酸中毒21 例临床和基因分析
2021-12-27 04:38张丽宁匡新宇康郁林朱光华黄文彦
临床儿科杂志 2021年12期
张丽宁 匡新宇 孙 蕾 冯 丹 康郁林 朱光华 黄文彦
上海市儿童医院 上海交通大学附属儿童医院肾脏风湿科(上海 200333)
原发性远端肾小管性酸中毒(distal renal tubular acidosis,dRTA)是由于远端肾小管泌H+功能障碍,尿液不能正常酸化,从而引起阴离子间隙正常的低血钾、高氯性代谢性酸中毒。患儿通常表现为生长迟缓、喂养困难、多饮多尿、高钙尿、髓质肾钙质沉着症、佝偻病或者骨软化等。本病具有遗传异质性,现已知多种基因变异与dRTA相关。随着基因诊断技术的进展,越来越多的疑难病例通过基因测序明确诊断。基因诊断也成为dRTA 明确诊断的有效工具。本文总结近5年收治的21例原发性dRTA患儿的临床资料及基因测序结果,以期探讨本病的临床特征及基因诊断价值。
1 临床资料
收集2014年1月至2019年1月上海市儿童医院收治的21例原发性dRTA患儿的临床资料及基因检测结果。患儿均符合原发性dRTA 诊断标准[1]:①高氯血症代谢性酸中毒,血HCO3-<21 mmol/L,阴离子间隙正常;②尿pH 值>5.5;③尿、血二氧化碳分压差值(U-BpCO2)<20 mmHg;④滤过HCO3-排泄分数一般正常或轻度增高;⑤排除继发性因素。
21例患儿中男性12例、女性9例。中位起病年龄9 月(四分位数范围2~24 个月),中位确诊年龄31 个月(四分位数范围24~54个月),确诊年龄以0~2岁以内最为常见(16例,76.2%)。仅1例患儿有家族史。
首发症状以生长发育迟缓最为常见(15 例,71.4%),其次是多饮多尿(9例,42.9%)、喂养困难(9例,42.9%),部分表现为乏力(5例)和反复泌尿道感染(3例)。辅助检查提示,21例(100%)患儿均存在肾髓质钙质沉积,14例(66.7%)四肢长骨摄片符合佝偻病表现,9 例存在肾囊性病(孤立性肾囊肿3 例,多发性肾囊肿3例,髓质海绵肾3例),2例为感音性耳聋。12例(57.1%)患儿确诊时存在蛋白尿,其中8例为肾小管性蛋白尿、3例混合性蛋白尿,1例肾小球性蛋白尿。酸中毒纠正后,除1 例肾小球性蛋白尿持续存在外,其余患儿的蛋白尿均消失。
患儿临床诊断明确后均同时予枸橼酸钠、枸橼酸钾口服液(新华医院配制,100 mL/10 g)。枸橼酸盐的中位需求量:0~1 岁3.95(2.18~5.50)mmol/(kg·d),1~2岁3.20(1.83~6.22)mmol/(kg·d),2~5岁3.10(2.08~3.88)mmol/(kg·d),>5岁1.90(0.83~ 2.00)mmol/(kg·d)。枸橼酸盐的最低需求量为0.83 mmol/(kg·d),为1 例13 岁男孩;最高需求量为 7.00 mmol/(kg·d),为1例1岁9月龄女孩。
为明确基因变异类型,经医院医学伦理委员会批准(No.2021 R 039-E 01),征得患儿监护人同意,10 例患儿行全外显子基因测序。抽取患儿及其父母的静脉血,送至晶能医疗科技(上海)有限公司,行Nimblegen 全外显子测序。采用Roche Nimblegen Exome Kit V4全外显子捕获芯片进行高通量测序,经Illumina NovaSeq 6000高通量测序仪完成测序(检测数据平均深度为100×以上)。利用BWA软件将测序结果比对到人类已知参考基因组序列hg 19(UCSC)中,用GATK3.1.1变异检测软件,采用ANNOVAR对变异位点进行注释。对筛选的可疑位点利用ABI 3730测序仪进行Sanger测序验证,并经序列分析软件得到验证结果。
10 例患儿中6 例检出5 种有意义的变异。例1为SLC4A1基因c.1765 C>T(p.R589C)新发变异,父母均为野生型。例2、3 均为SLC 4 A1基因c.2102 G>A(p.G701D)纯合变异,父母均为杂合型。例4为ATP6V1B1基因 c.687 G>A(p.P229G)、c.1397C>A(p.S466X)复合杂合变异,c.687G>A(p.P229G)为新发变异,父母均为野生型;c.1397C>A(p.S466X)为杂合变异,来源于患儿母亲,父亲为野生型。例5为ATP6V1B1基因c.232 G>A(p.G 78 R)、c.1354 del(F 452 fs)复合杂合变异,c.232 G>A(G 78 R)杂合变异来源于患儿父亲,母亲为野生型;c.1354 del(p.F 452 fs)杂合变异来源于患儿母亲,父亲为野生型。例6为ATP6V0A4基因c.1376C>T(p.S459P)、c.1029+5G>A的复合杂合变异,c.1376C>T杂合变异来源于患儿父亲,母亲为野生型;c.1029+5 G>A杂合变异来源于患儿母亲,父亲为野生型(表1,图1)。3例复合杂合变异目前均未见报道。
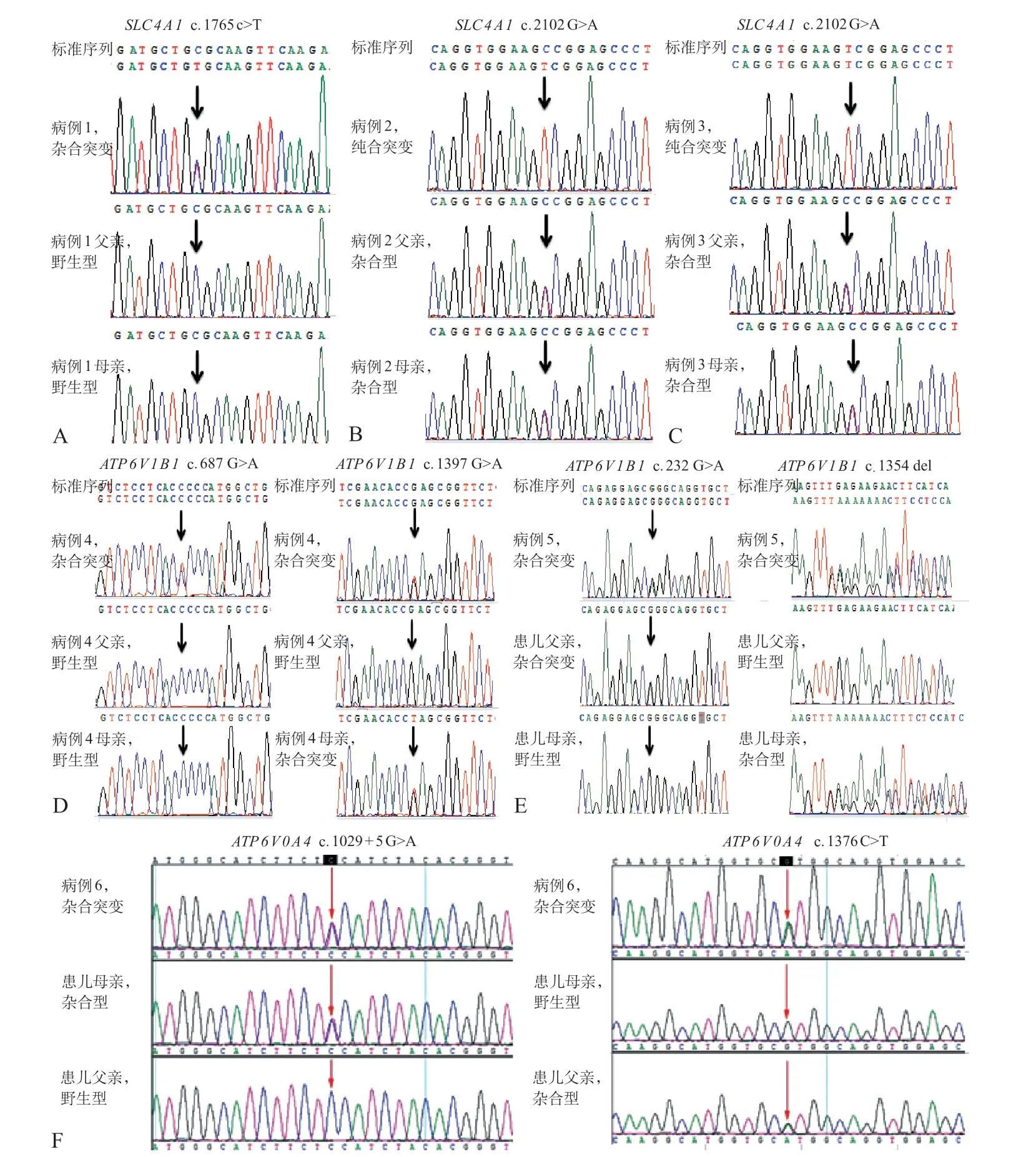
图1 6 例患儿DNA 序列图谱
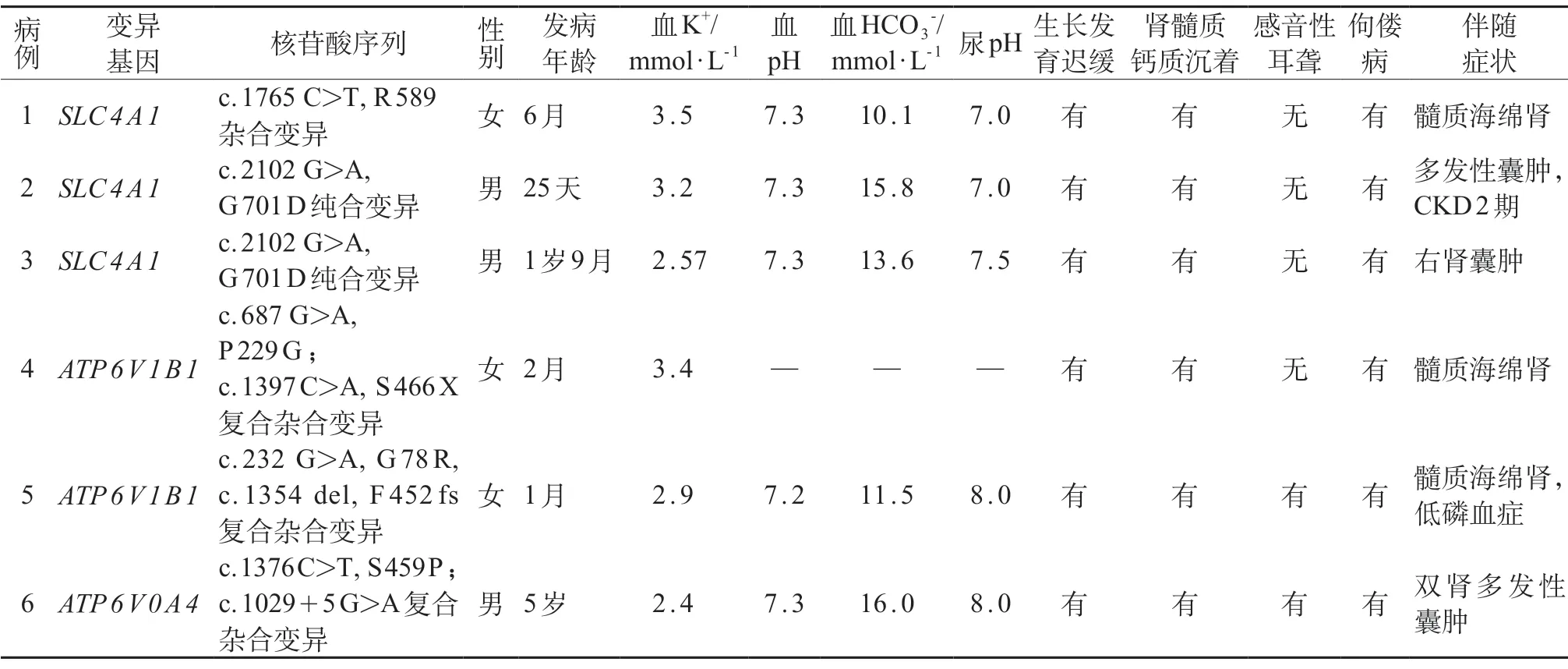
表1 6例患儿基因变异位点及临床表现
本组患儿随访14~126个月,中位随访时间45个月(28~61 个月)。所有患儿明确诊断后至末次随访时均遵医嘱规律服药。至末次随访时间,所有患儿血pH、血清碳酸氢盐、血清钾离子浓度、尿钙/肌酐均正常。大多数患儿身高达到正常水平,仅2 例身高低于同地区、同年龄、同性别身高的第3 百分位数。21 例患儿中仅2 例肾髓质钙质沉积完全消退,其余患儿可观察到钙质沉积的改善。仅1 例患儿血肌酐升高至65μmol/L,估算肾小球滤过率为75 mL/(min·1.73m2),为慢性肾脏病(chronic kidney disease,CKD)2期。
2 讨论
儿童原发性dRTA 为一种罕见的遗传异质性疾病,估计发病率<1:100000[2]。发病年龄为生后早期至青春期,临床表现复杂多样,常表现为生长发育迟缓、喂养困难、多饮多尿、反复泌尿道感染、乏力等,严重者可出现骨骼肌麻痹、心律失常等。实验室及影像学特征性表现是正常阴离子间隙的代谢性酸中毒、低钾血症、高尿pH 值、高钙尿症、肾脏钙质沉着症和
肾结石、佝偻病和骨软化症等。本组患儿以生长发育迟缓最为常见,其次是多饮多尿、喂养困难。所有患儿病初诊断时均存在阴离子间隙正常的代谢性酸中毒、碱性尿、高钙尿症,泌尿系统影像学均提示存在肾髓质钙质沉着症,这与既往文献报道相符[3]。值得注意的是,肾钙沉着症仅出现于髓质,如患儿表现为皮质型肾钙沉着症应及时考虑其他诊断,如原发性高草酸尿症[4]。本组患儿中9例存在肾囊性病,占42.8%,与既往报道的37.5%[5]大致相仿。囊肿表现为单纯性肾囊肿至多发性肾囊肿、髓质海绵肾等不同程度的受累。发病机制目前尚不清楚,推测可能是由于低钾血症刺激囊肿衬里上皮细胞生长、增殖所致[6]。相关研究曾报道了2例ATP6V1B1和ATP6V0A4基因变异的dRTA 患者同时伴有肾髓质海绵肾,指出髓质海绵肾可能是ATP6V1B1和ATP6V0A4基因变异所致dRTA 临床表型的一部分[7]。本组患儿中3 例髓质海绵肾最初均因影像学检查而被误诊,最终由基因检测明确诊断。其中1 例为SLC4A1基因变异,2例ATP6V1B1基因变异,提示SLC4A1基因变异的dRTA 患儿也可伴随髓质海绵肾,由此推测髓质海绵肾与基因型之间可能无特异性对应关系。
除上述典型的dRTA的临床表现外,本组有12例(57.1%)患儿确诊时存在蛋白尿,其中8例肾小管性蛋白尿,1 例肾小球性蛋白尿,3 例混合性蛋白尿。目前临床上几项关于dRTA 儿童出现部分Fanconi 综合征(低分子蛋白尿、高磷尿、氨基酸尿)的报道发现,在治疗开始后,所有儿童的近端肾小管病变都消失 了[5,8]。本组患儿酸中毒纠正后所有肾小管性蛋白尿均消失,与上述报道相符。值得注意的是,本研究中有4例患儿存在肾小球性蛋白尿。已有研究发现低钾性肾小管疾病患儿的肾小球和肾小管性尿微量蛋白普遍升高,指出低钾性肾小管疾病不仅累及肾小管,肾小球同样容易受损[9],但其分子机制有待进一步研究,有推测可能与肾素-血管紧张素-醛固酮系统的激活有关,也有学者认为与长期低血钾相关[10]。
儿童原发性dRTA治疗的目标是纠正代谢性酸中毒和其他生化异常,预防肾钙质沉着症和慢性肾脏病等并发症。目前唯一可用且有效的治疗方法是碱替代,治疗原则为早期、足量、终生服用。国内最常用碱剂为枸橼酸盐合剂,需求量遵从年龄越小需求量越高的规律[8]。本组患儿明确诊断后均予以枸橼酸钠、枸橼酸钾口服液联合治疗,碱需求量符合上述规律,但存在个体间差异。至末次随访时间,本组所有患儿代谢紊乱均得到了控制。大多数患儿出现了追赶性生长,仅2 例患儿身高仍低于同地区、同年龄、同性别身高的第3 百分位数,此2 例患儿从发病至确诊时间较长(1 例28 个月,1 例30 个月)而延误了治疗。1 例患儿在病程第6年时进展至CKD 2期。长期以来原发性dRTA 被认为是一种很少引起肾功能损害的良性疾病,然而近年来多项研究发现,18 岁以内儿童原发性dRTA患者CKD的发生率并不低,分别为37.5%、31.3%、28.8%,均为CKD 2、3期[5,11-12]。这种轻、中度肾损害的病因尚不清楚,有研究认为延误诊治、反复脱水和急性肾损伤、肾钙化或肾结石、慢性低钾血症、肾髓质囊肿、反复发作的急性肾盂肾炎均是潜在的危险因素[13],因此临床医师应加强对患儿肾功能的监测和随访。本组患儿较低的CKD 发病率可能归因于较短的随访期。
原发性dRTA 是遗传异质性疾病,基因检测越来越多应用于临床诊断及遗传咨询。目前6 种基因,ATP6V1B1、ATP6V0A4、SLC4A1、FOXI1、WDR72和ATP6V1C2,被认为是dRTA 的致病基因[14]。其中以ATP6V1B1、ATP6V0A4和SLC4A1基因的变异为主,占总检出率的58%~70%[14]。ATP6V1B1、ATP6V0A4基因变异均可导致H+-ATP酶结构或功能异常,致使远曲小管上皮细胞泌H+功能障碍,从而引起dRTA。因这两种基因也表达于耳蜗内淋巴囊的上皮细胞中,因此可引起进行性感音神经性聋和前庭导水管异常增大[3]。ATP6V1B1和ATP6V0A4基因变异的遗传方式均为常染色体隐性遗传。本组患儿中例4为ATP6V1B1基因 c.687 G>A(p.P229G)、c.1397C>A(p.S466X)复合杂合变异,理论上c.687G>A(p.P229G)为同义变异,但由于该碱基位于第7 号外显子的最后一个碱基,可能对剪切有一定的影响;c.1397 C>A(p.S466X)为无义变异,根据美国医学遗传学与基因组学学会(the American college of medical genetics and genomics,ACMG)判断标准为致病性变异。该患儿2个月时发病,脑干听力诱发电位目前无异常,需继续随访观察。例5为ATP6V1B1基因c.232 G>A(p.G78R)、c.1354 del(p.F452fs)复合杂合变异,根据ACMG判断标准,c.232 G>A(p.G78R)为致病性变异,c.1354 del(p.F452fs)为临床意义未明的变异。该患儿12个月发病,31个月确诊,确诊时即存在听力异常。例6为ATP6V0A4基因c.1376C>T(p.S459P)、c.1029+5G>A的复合杂合变异,根据ACMG 判断标准,2 个变异均为致病性变异。该患儿48个月时出现临床症状,72个月确诊,确诊时即存在严重低钾血症(2.4 mmol/L)、肾脏钙质沉着症、听力异常。上述3 例复合杂合变异均未见文献报道,根据ACMG 判断标准为致病性变异,但仍待进一步的功能验证。
SLC4A1基因编码位于肾小管上皮细胞基底膜外侧表面和红细胞膜上的Cl-/HCO3-交换体(AE1),为常染色体显性或隐性遗传[15]。常染色体显性遗传的方式更为常见,临床症状也相对温和。隐性遗传者发病年龄更早,临床症状重,除dRTA外,还可伴或不伴有血液系统疾病,如遗传性球形细胞增多症、东南亚卵圆细胞增多症等,多报道于东南亚地区[16]。本组患儿中病例1为SLC4A1基因c.1765 C>T(p.R589C)的新发变异,已有报道该变异为致病性[17],符合常染色体显性遗传规律。该患儿6个月起病,18个月确诊,无低钾血症,酸中毒症状轻(血pH 7.28),但也存在肾脏钙质沉着症、佝偻病表现。例2、例3 均为SLC4A1基因c.2102 G>A(p.G701D)纯合变异,父母均为杂合型且无临床症状,符合常染色体隐性遗传规律,已有报道其为致病性[18]。2 例患儿均为生后早期发病,伴有肾脏钙质沉着症、佝偻病和肾脏囊肿表现。例2 低钾血症、酸中毒程度轻(血钾3.2 mmol/L,pH 7.32),但至末次随访时间,该患儿已进展至CKD 2 期。例3低钾血症、酸中毒程度均较例2严重(血钾2.57 mmol/L,pH7.28),但无CKD的表现。2例患儿均无血液系统的表现。
本文介绍了儿童原发性dRTA 的临床表现,并在10例基因检测的患儿中发现2 例ATP6V1 B1复合杂合变异,1 例ATP6V0A4复合杂合变异,均未见临床报道,扩展了基因变异谱,有助于明确诊断和遗传 咨询。
猜你喜欢
湖南饲料(2021年4期)2021-10-13
现代畜牧科技(2021年4期)2021-07-21
种子(2021年3期)2021-04-12
健康之家(2020年15期)2020-05-08
中国医学创新(2019年9期)2019-08-19
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
滨州医学院学报(2016年2期)2016-05-27
中学生理科应试(2016年7期)2016-05-14
药品评价(2015年11期)2015-12-08
