
肉桂抗病毒有效部位总糖测定及单糖组成分析
2021-12-24 03:07郭明里王怀友张建文杨兆祥
亚太传统医药 2021年12期
李 滢,王 丽,郭明里*,李 宁,*,王怀友,张建文,杨兆祥
(1.昆药集团股份有限公司,云南 昆明 650100;2.香港科技大学深圳研究院 中药研发中心,广东 深圳 518057)
中药肉桂是樟科植物肉桂(CinnamomumcassiaPresl)的干燥树皮,能补火助阳,引火归元,散寒止痛,温通经脉[1]。肉桂是我国大宗常用中药材,同时也是世界闻名的著名香料植物,广泛应用于食品饮料、日用化妆品、香料工业[2]。因此从肉桂中寻求具有医药保健作用的活性分子及分子群不仅是中医药领域的热点,同时也是一个世界性的研究课题。
在中医药传统用法中,肉桂单方用于抗病毒治疗的报道不多见。但近年来国外学者报道肉桂的水溶性提取物具有显著的抗病毒活性[3-4]。该研究引起了我们的关注,我们通过以抗病毒活性为指导的追踪分离,获得了肉桂水溶性抗病毒有效部位KPC-rg1[5]。经反复验证,其抗病毒活性稳定且较国外学者获得的有效部位活性更高。KPC-rg1不但可用于各类疱疹病毒感染外用治疗性药物的开发,还有望用于包膜类病毒灭活疫苗的制备[5-6]。
KPC-rg1的化学结构目前尚不清晰。我们认为KPC-rg1是一类水溶性的酚性色素,由多酚及糖类物质经氧化聚合反应而形成,是一类结构复杂的异质的酚性化合物的总称。KPC-rg1兼有鞣质和糖类的部分理化性质[5]。目前尚无法从中分得单体化合物并鉴定结构。为逐步揭示其化学本质,并寻求其质量控制手段,我们进行了多方面的研究。采用中红外光谱技术结合化学计量学研究其指纹图谱,基于整体性和模糊性表征其宏观化学信息[7]。采用磷钼钨酸-干酪素法测定其中鞣质和酚性成分的含量[8]。本文从糖化学的角度,采用硫酸-苯酚比色法建立KPC-rg1总糖的含量测定方法。进而采用1-苯基-3-甲基-5-吡唑啉酮(PMP)柱前衍生化法对KPC-rg1进行单糖组成分析,明确其单糖种类和组成比例,并获得特征图谱。本研究为KPC-rg1的进一步研究、开发和综合利用提供技术手段并奠定实验基础,以期形成具有我国自主知识产权的中药、天然创新药物,为中药复杂体系有效部位新药开发提供参考。
1 仪器、材料与试剂
1.1 仪器
安捷伦1260高效液相色谱仪(包括在线脱气机、四元泵、自动进样器、DAD检测器)(美国Agilent公司);UV-2450紫外可见分光光度计(日本岛津公司);5510E-DTH超声仪(美国必能信公司);Milli-Q超纯水机(Millipore公司);CP225D电子分析天平(赛多利斯公司)。
1.2 材料与试剂
肉桂抗病毒有效部位(KPC-rg1)(批号:20150626,20151201,20160425)由昆药集团股份有限公司提供,制备工艺同文献[5-7]。阿拉伯糖、葡萄糖、甘露糖、半乳糖、葡萄糖醛酸对照品购自Alfa Aesar公司。鼠李糖对照品购自TCI公司。艾杜糖醛酸对照品购自Carbosynth公司。岩藻糖、半乳糖醛酸对照品及1-苯基-3-甲基-5-吡唑啉酮(PMP)购自Sigma-Aldrich公司。乙腈为色谱纯,购自德国默克股份两合公司。水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1 苯酚-硫酸分光光度法测定总糖含量
2.1.1 溶液制备 苯酚溶液:精密称取苯酚适量,加水配成浓度为5%的溶液,即得,置于冰箱中备用(现用现配)。葡萄糖对照品溶液:精密称取葡萄糖对照品适量,加水配成每1 mL含0.10 mg的溶液,即得。供试品溶液:精密称取KPC-rg1适量,加水配成每1 mL含0.28 mg的溶液,即得。
2.1.2 检测波长选择 精密吸取供试品和葡萄糖对照品溶液各1 mL,分别置于20 mL具塞刻度试管中,分别精密加入1 mL水,1 mL 5%苯酚溶液,室温下快速精密加入5 mL浓硫酸,冷却10 min,混匀后放置20 min。另取1 mL水同上操作制得空白溶液。紫外-可见分光光度计于波长400~600 nm范围进行扫描。供试品溶液和对照品溶液均在490 nm波长处有最大吸收,且无其它干扰峰影响分析,故选择490 nm作为检测波长。
2.1.3 标准曲线绘制 精密吸取葡萄糖对照品溶液0.4 mL、0.6 mL、0.8 mL、1 mL、1.2 mL、1.4 mL,分别置于20 mL具塞刻度试管中,精密加水至2.0 mL,精密加入1 mL 5%苯酚溶液,室温下快速精密加入5 mL浓硫酸,冷却10 min,混匀后放置20 min。另取相应体积的水同上操作制得相应的空白溶液。紫外-可见分光光度计于490 nm波长处下测定吸光度,每份测两次,计算平均值。以葡萄糖浓度为横坐标X,吸光度为纵坐标Y绘制标准曲线,得回归方程Y=53.059X-0.021 2,r=0.999 8。结果表明在0.005 0~0.017 5 mg·mL-1范围内,浓度与吸光度呈良好的线性关系。
2.1.4 精密度试验 精密吸取对照品溶液1 mL置于20 mL具塞刻度试管中,按“2.1.3标准曲线绘制”项下的方法,自“精密加水至2.0 mL”起,依法操作并测定吸光度,连续测6次,RSD为0.15%,结果表明仪器精密度良好。
2.1.5 重复性试验 取同一批号样品,按“2.1.1”项下的方法平行制备6份供试品溶液,分别精密吸取对照品溶液1 mL置于20 mL具塞刻度试管中,按“2.1.3标准曲线绘制”项下的方法,自“精密加水至2.0 mL”起,依法操作并测定吸光度,RSD为0.98%,结果表明方法重复性良好。
2.1.6 稳定性试验 精密吸取供试品溶液1 mL置于20 mL具塞刻度试管中,按“2.1.3标准曲线绘制”项下的方法,自“精密加水至2.0 mL”起,依法操作,于0、15、20、25、30、35、40、45、50、55、60 min测定吸光度,吸光度RSD为1.53%,结果表明溶液于60 min内稳定性良好。
2.1.7 加样回收率试验 分别精密称取已知含量的同一批号样品9份,每份14 mg,分别置于100 mL容量瓶中。分3组,每组3份,各分别加入50%、100%和150%的葡萄糖对照品,加水定容至刻度,摇匀,制得9份加样回收供试品溶液。分别精密吸取加样回收供试品溶液1 mL置于20 mL具塞刻度试管中,按“2.1.3标准曲线绘制”项下的方法,自“精密加水至2.0 mL”起,依法测定,计算加样回收率,平均回收率为99.25%,RSD为1.45%。结果表明本方法加样回收率良好。
2.1.8 样品含量测定 取3批样品,按“2.1.1”项下的方法制备供试品溶液,分别精密吸取供试品溶液1 mL置于20 mL具塞刻度试管中,按“2.1.3标准曲线绘制”项下的方法,自“精密加水至2.0 mL”起,依法测定,根据标准曲线计算各批次样品中的总糖含量,批号20150626、20151201、20160425样品的总糖含量分别为36.12%、39.23%和41.66%,平均值为39.00%。
2.2 PMP柱前衍生化法分析单糖组成
2.2.1 溶液制备 单糖对照品溶液:分别精密称取各单糖对照品适量,加水配成每1 mL含1.0 mg的溶液,使用时按需要以水稀释或混合配成单标或混合对照品溶液。
KPC-rg1样品酸水解及PMP衍生化:精密称取KPC-rg1适量,加水配成每1 mL含2.0 mg的溶液。精密量取该溶液300μL至5 mL具塞试管中,加入4 mol·L-1三氟乙酸300μL,充N2,封管,110 ℃水解4 h。水解液置70 μC水浴锅中蒸干,蒸干过程中加入甲醇适量以除去水解液中的三氟乙酸。蒸干后的样品加水50μL溶解,加入0.6 mol·L-1NaOH溶液50μL和0.5 mol·L-1PMP甲醇溶液100μL,涡旋混匀。置70 ℃烘箱反应60 min,取出室温放置10 min,加入0.3 mol·L-1HCl 100μL中和,加入三氯甲烷500 μL萃取3次,小心弃去氯仿层,上清液加水定容至1 mL。
单糖对照品的PMP衍生化:精密量取标准单糖对照品溶液50μL,不进行酸水解,PMP衍生化方法与样品一致,自“加入0.6 mol·L-1NaOH溶液50μL和0.5 mol·L-1PMP甲醇溶液100μL”开始如法操作。
2.2.2 色谱条件 Agilent Eclipse XDB-C18色谱柱(150 mm×4.6 mm,5μm);以0.1 mol·L-1乙酸铵(pH=5.5)缓冲液-乙腈(85∶15)为流动相进行等度洗脱;流速1 mL·min-1;柱温30 ℃;检测波长250 nm;进样量20μL。该色谱条件下,单糖混合对照品和样品PMP衍生物的HPLC见图1。
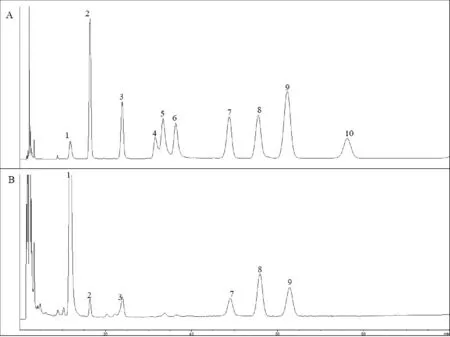
注:1:PMP;2:甘露糖;3:鼠李糖;4:葡萄糖醛酸;5:艾杜糖醛酸;6:半乳糖醛酸;7:葡萄糖;8:半乳糖;9:阿拉伯糖;10:岩藻糖。图1 样品PMP衍生物(A)和单糖混合对照品(B)的HPLC图
2.2.3 线性关系考察 对照图1单糖混合对照品和样品PMP衍生物色谱图中主要色谱峰的保留时间,确定KPC-rg1中的单糖主要由甘露糖、鼠李糖、葡萄糖、半乳糖和阿拉伯糖5种单糖组成。故针对该5种单糖进行方法学考察。
分别取甘露糖、鼠李糖、葡萄糖、半乳糖和阿拉伯糖对照品溶液,按“2.2.1”项下的规定衍生化,以水逐级稀释成一系列不同浓度的工作液,按“2.2.2”项下的规定进样分析,以浓度为横坐标X,峰面积为纵坐标Y绘制工作曲线,计算回归方程。结果见表1,各单糖在相应的质量浓度范围内线性关系良好。
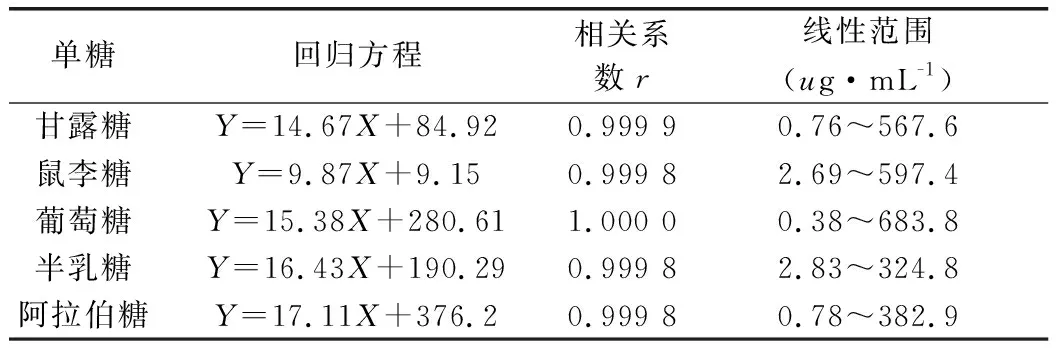
表1 5种单糖的线性关系
2.2.4 精密度试验 取KPC-rg1样品,按“2.2.1”项下的规定衍生化,按“2.2.2”项下的规定连续进样6次,记录峰面积。甘露糖、鼠李糖、葡萄糖、半乳糖和阿拉伯糖的峰面积RSD为0.21%~0.55%,表明仪器精密度良好。
2.2.5 重复性试验 取同一批号KPC-rg1样品,按“2.2.1”项下的规定平行制备6份衍生化样品,按“2.2.2”项下的规定进样分析,记录峰面积。甘露糖、鼠李糖、葡萄糖、半乳糖和阿拉伯糖的峰面积RSD为0.87~1.93%,表明方法重复性良好。
2.2.6 稳定性试验 取KPC-rg1样品,按“2.2.1”项下的规定衍生化,分别于0、2、4、6、8、12 h按“2.2.2”项下的规定进样分析,记录峰面积。甘露糖、鼠李糖、葡萄糖、半乳糖和阿拉伯糖的峰面积RSD为0.90%~1.89%,表明样品于12 h内稳定。
2.2.7 加样回收率试验 分别精密称取已知含量的同一批号样品6份,每份25 mg,分别置于25 mL容量瓶中,各分别加入100%的甘露糖、鼠李糖、葡萄糖、半乳糖和阿拉伯糖对照品,加水定容至刻度,摇匀,制得6份加样回收供试品溶液。分别按“2.2.1”项下的规定衍生化,按“2.2.2”项下的规定进样分析,计算加样回收率,甘露糖、鼠李糖、葡萄糖、半乳糖和阿拉伯糖的平均回收率为96.04%~101.35%,RSD为1.42%~1.97%,表明方法加样回收率良好。
2.2.8 样品单糖组成分析 取KPC-rg1样品,按“2.2.1”项下的规定衍生化,按“2.2.2”项下的规定进样分析,根据标准曲线计算各组成单糖含量。根据公式n=m/M(n为单糖摩尔数,m为单糖质量,M为单糖摩尔质量),计算各组成单糖的摩尔比例,甘露糖、鼠李糖、葡萄糖、半乳糖和阿拉伯糖比例为0.14∶0.28∶0.38∶1∶0.92。
3 讨论
KPC-rg1优秀的抗病毒活性和广阔的应用前景推动我们对其进行持续的研究开发。物质基础不明确是制约其成药性的关键问题。本课题组前期曾采用多种分离手段(包括硅胶、十八烷基键合硅胶、Sephadex LH-20等)尝试对KPC-rg1进行分离,但均未能获得单体化合物。随着研究的逐渐深入,我们认为KPC-rg1是一类水溶性、分子量不均一、复杂的天然多聚物,具有酚类和糖类的结构片段,经氧化、聚合、耦合等作用形成。类似的化合物在自然界广泛存在,较为著名的如茶褐素。茶褐素是普洱茶等黑茶中含有的水溶性高聚色素,颜色为棕褐色或棕红色,在普洱干茶中的含量可达12%,是构成黑茶品质特征的重要物质基础[9-10]。但由于结构的复杂性,有关其分离纯化及结构鉴定的研究至今仍处于探索中。该类复杂天然产物的结构特点为其质量控制提出了严峻的挑战。为此我们尝试针对其物理化学特征,从多个角度对其进行多维的表征。本研究针对其糖类结构片段进行了探索性研究。
苯酚-硫酸比色法是总糖含量测定的经典方法,该方法具有显色稳定、灵敏度较高、受其他碳水化合物和蛋白质干扰较小等优势,在中药、天然药物领域应用范围较为广泛[11-13]。本研究在方法建立过程中,针对KPC-rg1的特点对实验参数进行了细致的调整与优化,包括:苯酚浓度、苯酚用量、浓硫酸用量、浓硫酸加入方式、显色温度和时间等,从而确定了最适合的实验条件。本研究以葡糖糖为对照品对样品总糖含量进行测定。以葡糖糖计,3批KPC-rg1总糖含量的平均值为39.00%。作为测量值,因未考虑各个单糖的校正因子,该含量并非样品中糖类物质的真实含量,但可作为含量指标初步评价产品质量和一致性。
单糖组成分析是糖类结构表征和解析的重要手段。因糖大多没有发色团,单糖结构相近且极性较强,难以满足高灵敏度分析手段的检测要求。PMP衍生化方法可使糖类在245 nm处产生强烈的紫外吸收,条件较温和,无需酸催化剂,且产物无唾液酸异构,是较理想的衍生化方法[14,15]。本研究采用HPLC对PMP衍生产物进行分析检测,明确单糖种类和组成比例,并获得特征图谱。该方法在苯酚-硫酸比色法测定总糖含量的基础上,为KPC-rg1的质量评价和控制提供了更为精准的技术手段,并从糖化学的角度对KPC-rg1的物质基础进行了探索。
4 结论
本研究建立了肉桂抗病毒有效部位KPC-rg1总糖测定及单糖组成分析方法。实验结果表明该方法简便、快速、准确、可靠,可用于KPC-rg1的质量控制,为其进一步研究、开发和综合利用提供技术手段并奠定实验基础,为中药复杂体系有效部位新药开发提供了参考。
猜你喜欢
家庭百事通·健康一点通(2020年12期)2020-12-31
中国外汇(2019年13期)2019-10-10
中国外汇(2019年7期)2019-07-13
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
教育教学论坛(2018年6期)2018-03-15
中国医药导报(2017年31期)2017-12-19
中国中药杂志(2017年20期)2017-11-11
科技资讯(2017年20期)2017-08-22
青春岁月(2016年24期)2017-04-15
