
利用CRISPR/Cas9技术进行lncRNA snhg17-KO基因敲除小鼠模型的构建与鉴定*
2021-12-23 07:15洪马林
医学理论与实践 2021年23期
许 静 高 琳 洪马林 赵 盼
暨南大学第二临床医学院 深圳市人民医院 1 病理科 2 临床医学研究中心,广东省深圳市 518000
随着对人类基因组研究数据的不断探索发现,非编码RNA在整个基因组中占据98%的作用,长链非编码RNA简称lncRNA,是一种长度>200个核苷酸,但不具有蛋白质编码功能的序列RNA。研究表明,lncRNA作用于细胞的增殖、分化、凋亡及免疫功能,并且与各种肿瘤密切相关,参与肿瘤的发生、发展过程[1-2]。一类长度在60~300个核苷酸,表达在核内的小核RNA(snRNA),常存于宿主基因内含子中,发挥着抑制或促进肿瘤细胞增殖的功能。而小核RNA宿主基因17(Small nucleolar RNA host gene 17,snhg17)作为一种重要的lncRNA,在肺癌、胃癌、结肠癌等多种肿瘤组织表达上调,且与临床预后及分期显著相关[3-5]。迄今为止对于snhg17在乳腺恶性肿瘤的具体分子机制,尚未见相关报告。因此探讨snhg17的功能,对于发现乳腺肿瘤特异性标志物及治疗预后具有深远的研究意义。
动物模型作为一种非常实用的实验研究工具,可以模拟出肿瘤的免疫表型,因此构建snhg17-KO基因敲除小鼠模型,对于后续实验研究具体机制及探寻肿瘤治疗至关重要。使用细胞生物手段解析靶基因功能的基因编辑技术,其中CRISPR/Cas9系统,具有高效、简单的优势,被实验室广泛应用,该项技术包括两个重要步骤:一是Cas9蛋白执行剪切功能;二是guide RNA(gRNA)特异性将Cas9蛋白引导至靶DNA序列[6-7]。我们让gRNA与Cas9蛋白结合构成复合物,使用gRNA引导并识别目的基因靶点,再进行Cas9蛋白对目标序列的切割。本研究拟通过选取snhg17基因1-6号的外显子,进行gRNA条件性有效敲除C57BL/6小鼠基因组中的snhg17基因,以达到构建snhg17-KO基因敲除小鼠模型的目的,为后续深入探讨snhg17在肿瘤中的分子机制提供优质的实验动物模型。
1 材料与方法
1.1 实验动物及主要试剂 C57BL/6小鼠购于北京维通利华实验动物技术公司,动物实验经由深圳市人民医院动物委员会批准,饲养于深圳市人民医院SPF级实验动物中心,实验分组:实验组小鼠(KO组)、对照组小鼠(CON组)、未注射的空白组小鼠(WT组)。M16培养基购于德国MERCH公司;pRP[CRISPR]-hCas9-U6载体购于美国Addgene公司,PCR引物及探针由上海生工合成;提取总RNA试剂、Lipofectamine 2000转染试剂盒及Omega试剂盒购于Invitrogen公司;pEASY-BLUNT Zero Cloning Kit载体连接试剂盒和Trans1-T1感受态细胞购于Trangene公司;DNA Pst I限制性核酸内切酶、高保真DNA聚合酶FastPfu购于Takara公司;0.5 M EDTA、3 M醋酸钠购于Thermo公司。
1.2 方法
1.2.1 载体设计及构建:依据CRISPR/Cas9技术 gRNA设计原理,采用数据库(https://zlab.bio/guide-design-resources)设计2对用于CRISPR/Cas9技术的gRNA,对位于小鼠2号染色体上的snhg17基因(Gene ID:68108)第3和第6外显子作为靶点,通过比较评分,选出效率高的gRNA3和gRNA4序列(表1)。构建至pRP[CRISPR]-hCas9-U6载体中,通过VectorBuilder构建带有Cas9质粒的融合质粒进行质粒构建。

表1 sgRNA序列
1.2.2 质粒转化与提取:将0.1μl的质粒与DH5α 42°C水浴混匀,取900μl LB培养基, 37℃45min震荡培养,使菌体复苏,取20μl菌体种板,37°C培养16h,筛选单克隆菌落,加4μl 100μg/ml Amp培养,用于质粒提取。采用Omega试剂盒在5ml LB培养基中加入目标质粒的E.coli,37℃振荡培养,取1.5ml菌液加入250μl(含RNase A)溶液,得到裂解液,离心取上清,与0.5倍100%乙醇混合,至2ml吸收柱反复清洗,滴加50μl无菌水在滤膜上离心。DNA检测:分光光度计检测,OD 260/280=1.8。
1.2.3 体外转录模板扩增及转录: 设计合成扩增引物(表2,下划线示T7启动子区域)。取pRP[CRISPR]-hCas9-U6质粒20ng,加入无酶双纯水50μl,含Mg2+扩增缓冲液25μl,dNTP1μl,DNA聚合酶1μl,上游及下游引物1μl,所有操作均在冰上进行。PCR反应程序:预变性95℃ 30min,变性95℃ 15s, 退火65℃15s,延伸72℃ 30s,35个循环;72℃ 5min。在反应体系液中加入PCR扩增产物,37℃温育1h,使用1μl TURBO DNase,1μl 0.5M EDTA进行体外转录并纯化,得到分离的gRNA3、gRNA4和Cas9蛋白的mRNA。
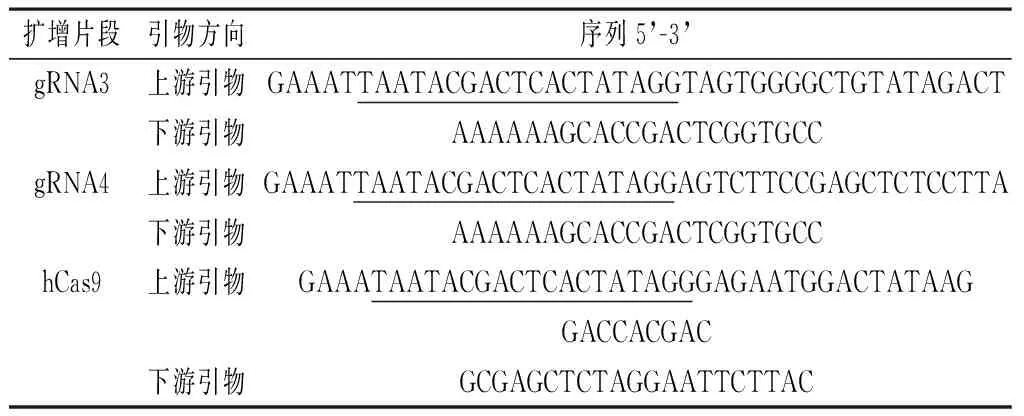
表2 体外转录模板扩增的引物序列
1.2.4 构建动物模型:(1)小鼠饲养:所有实验分组小鼠均饲养在深圳市人民医院清洁SPF级标准的中心实验室动物房,保持室温25℃,50%~60%湿度值,光照和黑暗12h交替循环,每日自由采食,垫料、水、饲料均经高压高温灭菌处理供给。(2)取受精卵:选取12周C57BL/6雌鼠诱导排卵,腹部注射促性腺激素(PMSG)120U/kg,与雄鼠交配,使用透明质酸酶消化剪取输卵管组织,显微镜下收集受精卵,37℃,5% CO2M16培养基培养。(3)显微注射:收集和培养C57BL/6小鼠受精卵,将注射针吸入gRNA3/Cas9和gRNA4/Cas9转录产物约0.5μl,插入卵核仁内注射。(4)植入代孕鼠:将受精卵植入代孕小鼠生殖系统,进行筛选并繁育F0代小鼠。
1.2.5 繁育snhg17杂合和纯合突变小鼠:待转染gRNA3与gRNA4的受精卵小鼠出生成熟后,再与WT小鼠(野生型)繁殖,得到的即为F1代。通过基因型鉴定出的F1代杂合子基因敲除小鼠,雌雄交配繁殖出F2代,再进行基因鉴定,最终得到两条染色体均带有snhg17突变基因的纯合突变小鼠。
1.2.6 小鼠基因型鉴定:剪取鼠尾并破碎,提取DNA使用TaKaRa MiniBEST 全基因组DNA提取试剂盒,PCR进行扩增目的片段同时行目的条带的电泳检测。PCR反应体系:加入1.5μl鼠尾基因组DNA,1.5μl dNTP,1.0μl Mg+缓冲液,0.5μl引物混合液,0.5μl DNA聚合酶,21.8μl ddH2O。PCR反应条件:预变性94℃ 5min;变性94℃ 30s;退火60℃ 30s;延伸72℃ 60s;循环35次,72℃再次延伸5min。为进一步验证,进行产物基因测序。
1.3 统计学方法 采用SPSS22.0软件进行统计学分析,采用独立样本t检验比较组间均数,以α=0.05为检验水准,P<0.05为差异有统计学意义。
2 结果
2.1 小鼠snhg17 gRNA构建与筛选 使用张峰实验室(https://zlab.bio/guide-design-resources)数据库,设计gRNA, snhg17位于小鼠2号常染色体上,已确定有6个外显子(exon),选择exon 1~exon 6区作为目标区域(图1所示)。设计2对评分较高的gRNA:gRNA1和gRNA2、gRNA3和gRNA4,最终选择效率高的gRNA3和gRNA4进行基因打靶。
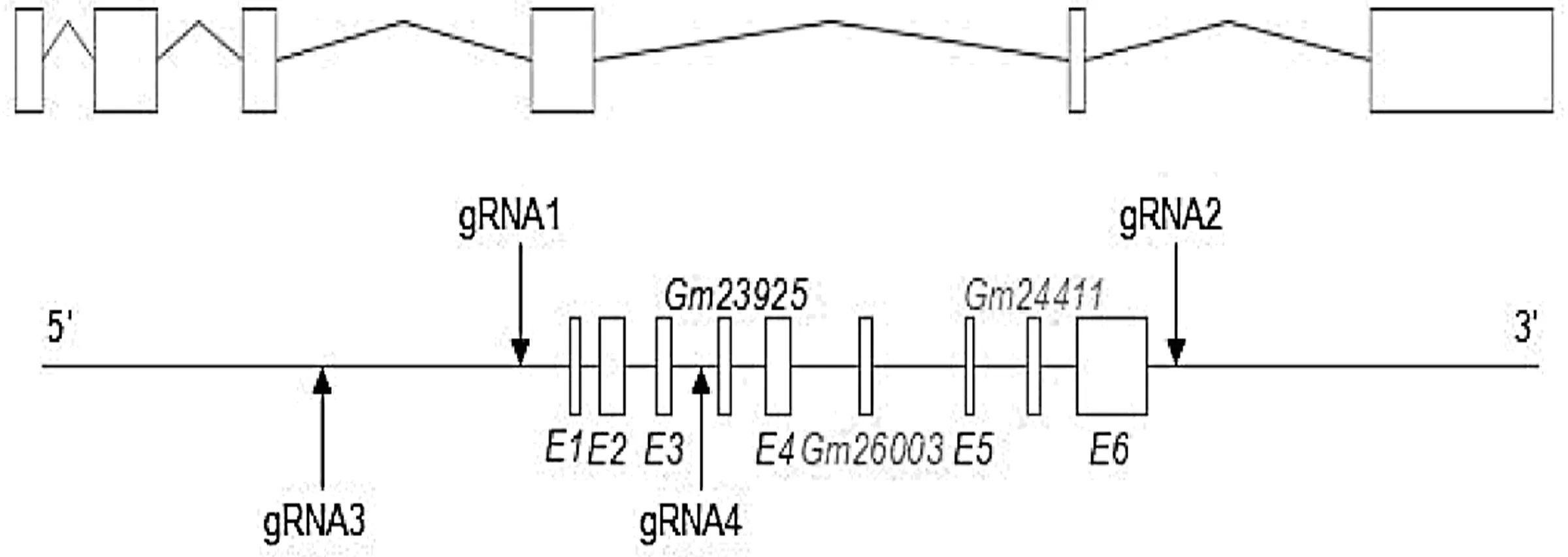
图1 gRNA设计策略
2.2 F1代snhg17杂合子小鼠的构建及基因型鉴定 在gRNA3和gRNA4区域两侧设计两对引物,用于小鼠的基因型测定(图2a)。F0代与WT小鼠繁殖得到的F1代,用于提取鼠尾DNA,PCR扩增电泳显示目的条带。图2b表明snhg17 gRNA是否打靶成功,如图所示,图中3、6、7、8、9、17、18、19号小鼠均打靶成功,敲除了目的片段。图2c表明snhg17 gRNA打靶成功,图中3、6、7、8、9、17、18、19号小鼠的电泳结果中显示出2条带,分别为650bp和437bp,表明杂合子敲除小鼠打靶成功,而WT小鼠仅有437bp处的一条带。图2d表明有4 202bp的基因被敲除,符合预期gRNA3和gRNA4的打靶条带大小。

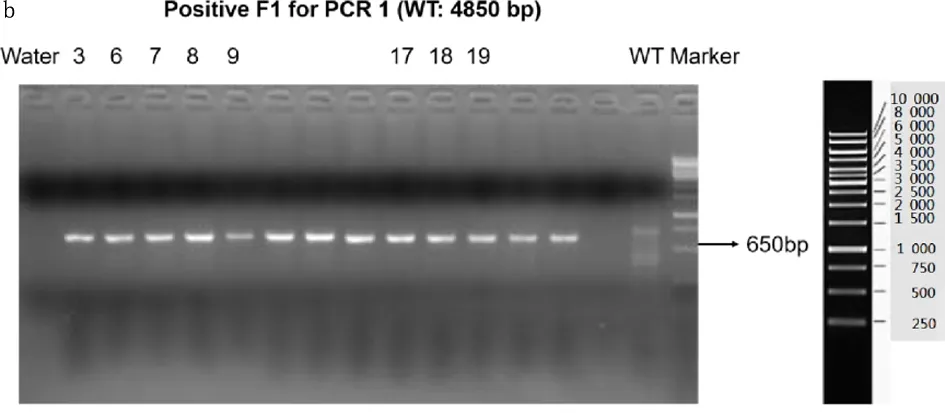
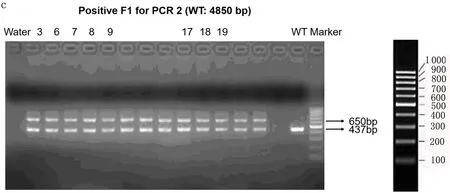
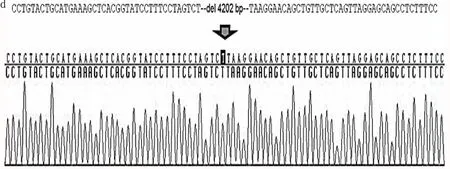
图2 snhg17基因敲除杂合子小鼠的验证a.gRNA设计策略 b.DNA电泳检测snhg17敲除小鼠的基因型情况 c.DNA电泳检测snhg17敲除小鼠的基因型情况 d.检测目的基因打靶效果
2.3 F2代snhg17纯合子小鼠的构建及基因型鉴定 设计4对引物用于验证F2代的基因型(图3a示)。PCR扩增电泳显示目的条带。图3b表明snhg17 gRNA是否打靶成功,其中8号小鼠打靶成功,敲除了目的片段。图3c表明snhg17 gRNA打靶成功,8号小鼠的电泳结果中显示仅有650bp 处有1条带,表明纯合子小鼠敲除打靶成功,而杂合子在840bp和650bp处各有一条带,WT小鼠在840bp处有一条带。图3d中8号小鼠的电泳结果中显示仅有650bp 处有一条带,表明纯合子敲除小鼠打靶成功,而杂合子在437bp和650bp处各有一条带,WT小鼠在437bp处有一条带。图3e中8号小鼠在694bp处没有条带,表明纯合子敲除小鼠打靶成功,而在694bp处有条带的小鼠则为杂合子或WT小鼠。图3f表明有4 202bp的基因被敲除,符合预期gRNA3和gRNA4的打靶条带大小。
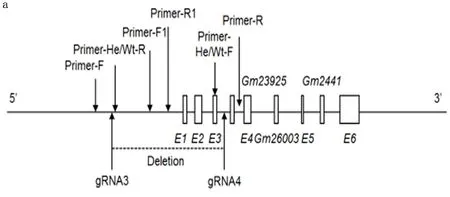

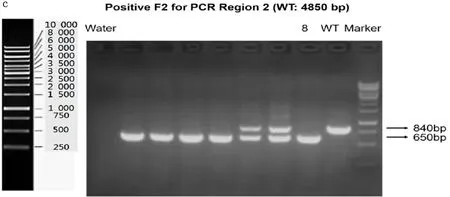
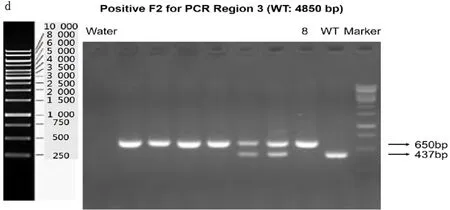
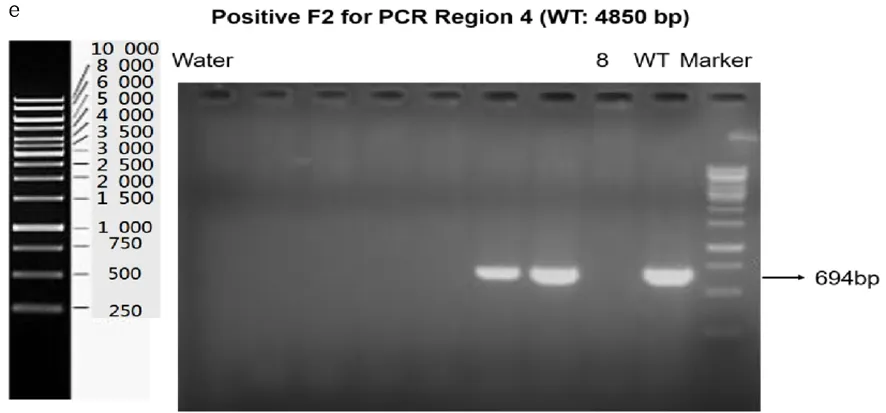
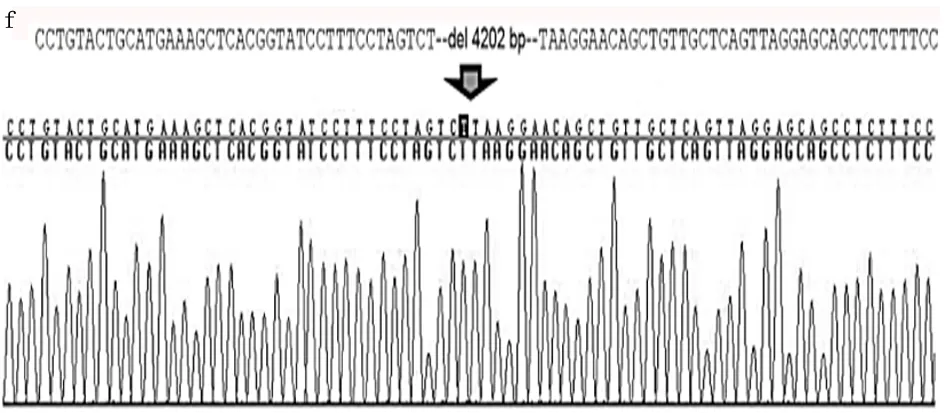
图3 snhg17基因敲除纯合子小鼠的验证a.验证引物设计策略 b.DNA电泳检测PCR region 1目的片段扩增情况 c.DNA电泳检测PCR region 2目的片段扩增情况 d.DNA电泳检测PCR region 3目的片段扩增情况 e.DNA电泳检测PCR region 4目的片段扩增情况 f.检测目的基因打靶效果
3 讨论
随着分子生物基因测序的应用,逐步建立起大量生物信息数据库,从而加速了对lncRNA这类非编码RNA作用机制的认识。lncRNA长度>200核苷酸,研究表明其缺乏开放性阅读框,但在5’端可加帽、剪接,3’端可被聚腺苷酸化, RNA聚合酶Ⅱ可以被转录,因此lncRNA被认为是一种结构性RNA、分子支架,参与并调节细胞遗传方面的功能,同时也是调控着miRNA的“分子海绵”[8]。
存在于核内的snoRNA,长度为60~300核苷酸,其宿主基因为lncRNA,在肿瘤进展中起到抑制或促进功能。snhg17基因位于20号染色体p12上,是这些宿主基因中重要的一员[9]。已被证实,在肺癌、胃癌中的表达上调,并且通过表观遗传方式,进一步调节了p15及 p57蛋白,达到促进肿瘤细胞的增殖及侵袭[3],另外在肠道肿瘤、乳腺肿瘤及恶性黑色素瘤中发挥致癌活性[10-12],然而目前对于其具体分子机制尚不十分明了。通过对人的胃癌组织进行转录组测序后发现,snhg众多亚型中snhg2与snhg5在肿瘤组织中的表达量下调,而snhg17的转录水平上调,证明snhg17参与胃肿瘤的发生发展[13]。此外,有研究证明乳腺癌患者肿瘤组织snhg17的耗竭抑制了体外细胞增殖、迁移和侵袭,并抑制了异种移植肿瘤模型中的肿瘤生长。snhg17可以充当BC细胞中miR-124-3p的内源海绵,通过海绵miR-124-3p调节BC的进程[12]。因此,深入研究snhg17在肿瘤中的作用机制非常重要,开发新型靶点药物对临床肿瘤治疗提供一种探索性科学理论依据。
由于小鼠与人类基因组有99%同源,同时易于引导特定基因的定向改变,允许基因精确的改变,因此采用建立动物模型的方式对临床疾病尤其肿瘤方面的机制探索具有重要意义。目前进行基因敲除小鼠的构建主要通过两种方法,一使用同源重组代替内源基因实现敲除目的,或者通过启动子/polyA捕获实现敲除。常用的技术手段有Cre/loxP、CRISPR/Cas9、ZFN和TALEN技术[14]。Cre/loxP系统是通过在靶位点两端设计loxP序列,经Cre酶处理后,两者方向相同,目的基因会被删除;两者相反则会被置换。但Cre/loxP技术在应用于基因工程小鼠模型上时价格昂贵并且耗时;ZFN技术特异性切割目的基因序列引起双链DNA断裂,从而启动了 DNA修复机制,进行定向修复;而TALEN技术是根据靶基因序列变换双氨基酸残基(RVDs),由转录激活因子样效应物(TALE)与靶点结合,被FokI切割以完成基因定点敲除。此外,TALEN结合靶基因序列更严格,靶向效率高,同时也能够降低脱靶效应等。然而,TALEN更适合于针对蛋白编码基因基因敲除,对于非编码基因并不适用。
CRISPR/Cas9技术价格经济,而且基因敲除长达20kb,有研究表明,在原发性肉瘤中将CRISPR/Cas用于建立基因工程小鼠模型后与Cre/loxP技术相比,CRISPR/Cas9能够更快地使细胞突变,在可控时间和空间下迅速产生并引发肉瘤形成,得到可靠的基因编辑小鼠[15],因此通过CRISPR/Cas9技术达到了敲除snhg17基因的目的。在本实验研究中使用体外转录产物而没有采用质粒的原因:由于质粒载体比较稳定,若直接注射质粒载体到受精卵中,则会在小鼠受精卵至胚胎时期存在较长时间,这样会导致在不同时期切割而产生嵌合体,不利于后期基因分型,另外质粒还有可能随机整合在基因组中。而注射转录后产物较不稳定,会在受精卵时期逐步降解,减少嵌合体的产生,方便后期基因分型,因此实验选择体外转录后注射方式。
综上所述,本研究应用CRISPR/Cas9技术在C57BL/6小鼠中敲除snhg17,通过PCR及测序基因型鉴定,成功构建了snhg17基因敲除小鼠,为探寻snhg17在肿瘤发生发展中的作用机制研究提供可靠的动物模型。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
计算机应用与软件(2022年6期)2022-07-12
表面工程与再制造(2022年2期)2022-06-02
自然灾害学报(2022年2期)2022-05-10
中国学校体育(2021年10期)2021-04-26
三农资讯半月报(2020年11期)2020-06-21
汽车实用技术(2019年21期)2019-11-22
江苏农业学报(2019年1期)2019-09-10
人民交通(2009年5期)2009-06-01
