
食品中米酵菌酸和毒黄素一测多评方法的探讨
2021-12-21 10:17叶文芳陈嘉聪王晓琴张丰芸黄秀丽朱文娟赵智锋
食品与药品 2021年6期
叶文芳,陈嘉聪,王晓琴,张丰芸,黄秀丽,朱文娟,赵智锋
(惠州市食品药品检验所,广东 惠州 516000)
近年,由椰毒假单胞菌酵米面亚种(Pseudomonas cocovenenans subsp. farinofermentans,简称椰酵假单胞菌)引起的食物中毒事件时有发生。据文献报道,椰酵假单胞菌在适宜的条件下会产生米酵菌酸(bongkrekic acid)和毒黄素(toxoflavin)[1-3]。其中,米酵菌酸毒性强于毒黄素,是主要致死因子。毒黄素毒性虽弱于米酵菌酸,但毒黄素会造成组织与细胞损伤,同时毒黄素还具有致突变作用和潜在的致癌性[4-6]。
一测多评法(quantitative analysis of multicomponents by single marker,QAMS)是利用易得、廉价、有效的化学物为代表性成分,借助待测物中有效成分的内在函数和比例关系,以一个标准品为参照物,建立该成分与其他成分间的相对校正因子,并通过其计算含量,使其计算值与实测值符合定量方法学的要求,实现多组分的同时测定[7]。本研究选取米酵菌酸为内参物,考察米酵菌酸与毒黄素之间的相对校正因子,探讨QAMS用于毒黄素含量测定的可行性。
1 仪器与材料
1.1 仪器
LC-20A高效液相色谱仪,配DAD 检测器(日本岛津公司);Agilent 1200 高效液相色谱仪,配DAD 检测器(美国安捷伦公司);Agilent 1260高效液相色谱仪,配DAD 检测器(美国安捷伦公司);Fotector Plus高通量全自动固相萃取仪(厦门睿科)。
1.2 材料
米酵菌酸(纯度≥95 %,上海安谱);毒黄素(纯度≥98 %,上海甄准);增强型脂质去除净化管(QuEChERS dSPE EMR-Lipid,广州安捷伦);除脂萃取盐包(Polish Tube-NaCl/MgSO4,广州安捷伦科);固相萃取净化管(dSPE Cleaneup Tubes,成分配比:MgSO4300 mg,PSA 100 mg,C18100 mg,容量15 ml,上海安谱);Cleanert®MAS-Q系列酸性物质净化固相萃取柱(成分配比:PSA 400 mg,C18400 mg,MgSO41.2 g,容量15 ml,天津博纳艾杰尔);甲醇、乙腈为色谱纯,其余试剂为分析纯;实验用水为超纯水;分析样品湿米粉、银耳、黑木耳、玉米面均为本实验室日常抽检的样品。
2 方法
2.1 色谱条件
色谱柱:C18色谱柱(柱长250 mm,内径4.6 mm,粒度5 μm);流动相:甲醇(A)和水(B),水用甲酸调pH 3.0;流速:1 ml/min;检测波长:258 nm;柱温:30 ℃;进样量:10 µl,梯度洗脱程序见表1。

表1 毒黄素和米酵菌酸的测定梯度洗脱程序
2.2 对照品溶液配制
准确称取米酵菌酸对照品10 mg,加甲醇溶解,转移至100 ml量瓶中,加甲醇定容;准确称取毒黄素对照品10 mg,加甲醇溶解,转移至100 ml量瓶中,加甲醇定容。准确吸取上述溶液各25 ml,置于同一50 ml量瓶中,配制成50 µg/ml的混合对照品储备液。分别取混合对照品储备液适量,配制成毒黄素、米酵菌酸浓度为0.5,1.0,2.0,5.0,10.0,25.0 µg/ml的系列混合对照品溶液。
2.3 样品前处理
取粉碎混匀的试样约2 g,精密称定,于50 ml塑料离心管中,加入甲醇-水(8:2,v/v)10 ml,涡旋2 min,超声10 min,5000 r/min离心5 min,吸取上清至预装dSPE EMR-Lipid吸附剂的离心管中,涡旋2 min,5000 r/min离心5 min,然后取上清于氮吹管中;重复上述提取、净化步骤,合并上清后于40 ℃水浴中氮吹至低于1 ml,用甲醇定容至1 ml,0.22 μm滤膜过滤后进行分析。
3 结果与分析
3.1 提取方法及色谱条件的优化
以湿米粉为基质,考察不同提取溶剂(甲醇、乙腈、80 %甲醇、80 %乙腈),提取次数,流动相pH值(pH 2.5,pH 3.0,pH 3.5)及其酸度调节剂(甲酸、乙酸、磷酸),以及检测波长对毒黄素及米酵菌回收率的影响。结果表明,以80 %甲醇为提取溶剂,提取2次,以甲酸作为流动相B的酸度调节剂,流动相B pH 3.0及检测波长258 nm时,毒黄素和米酵菌酸的回收率均最优。
3.2 方法学考察
3.2.1 线性关系、方法检出限及定量限 分别精密吸取2.2项下系列浓度的混合对照品溶液各10 µl,按2.1项下色谱条件进样分析。以峰面积为纵坐标,米酵菌酸和毒黄素浓度为横坐标,绘制标准曲线,进行线性回归。结果表明,米酵菌酸和毒黄素在0.5~25 µg/ml范围内呈良好的线性关系,相关系数分别为0.99999和0.99991。线性关系、方法检出限及定量限见表2。

表2 米酵菌酸和毒黄素的线性关系、方法检出限及定量限
3.2.2 稳定性试验 取湿米粉样品,按2.3项方法制备供试品溶液,室温放置,分别于0,6,12,18,24,30,36 h进样10 µl,测定,测得18 h内毒黄素峰面积RSD为4.79 %,而后峰面积明显衰减,24,30,36 h的RSD分别为6.13 %,7.46 %,8.95 %,RSD均大于5 %;36 h内米酵菌酸峰面积RSD为0.83 %,稳定性较好。
3.2.3 方法回收率与精密度 选取银耳、黑木耳、玉米面及湿米粉4种不同基质,进行3水平的加标回收试验和精密度试验, 每种基质的每个水平进行6次平行测定,结果见表3。在2,8,16 μg/ml 3个加标水平下,毒黄素的平均回收率为88.94 %,RSD为1.50 %;米酵菌酸的平均回收率为90.43 %,RSD为1.43 %。结果表明方法的准确度和精密度良好。
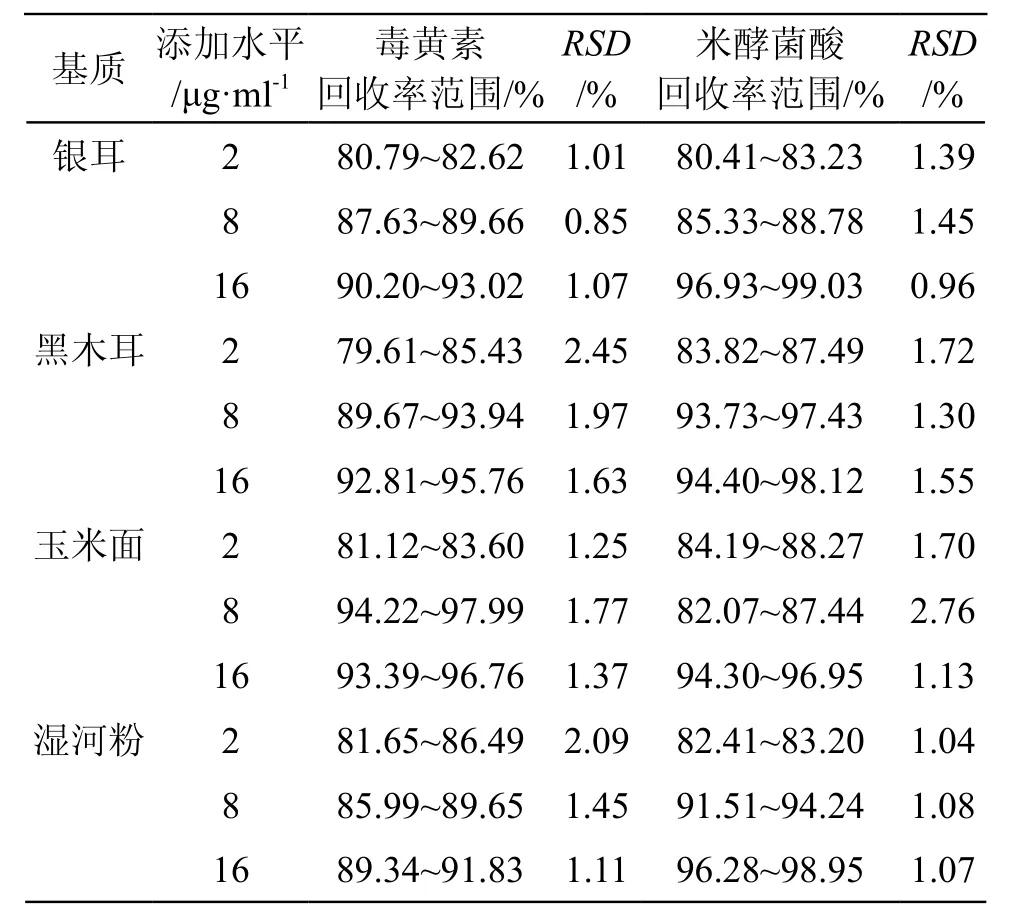
表3 毒黄素和米酵菌酸的回收率结果(n=6)
4 相对校正因子的建立及其适用性评价
4.1 相对校正因子(f)的建立
精密吸取2.2.4项下混合对照品工作溶液,分别取1,2,5,10,15,20 µl,注入液相色谱仪,记录峰面积,以米酵菌酸为内参物,按下式计算QAMSf值,结果见表4。

表4 f值测定结果

式中Ci为内标物浓度,Ai为内标物峰面积,Cs为组分s浓度,As为组分s峰面积)[7-8]。
4.2 相对保留时间(RT)的确定
RT指各待测成分与内参物间保留时间的比值。结果表明待测组分间的RT值波动较小(RSD<5 %),因此在缺乏对照品的情况下,利用RT值,并根据内参物的保留时间,结合色谱峰的紫外吸收特征,能比较准确合理地确定目标成分的位置。结果见表5。
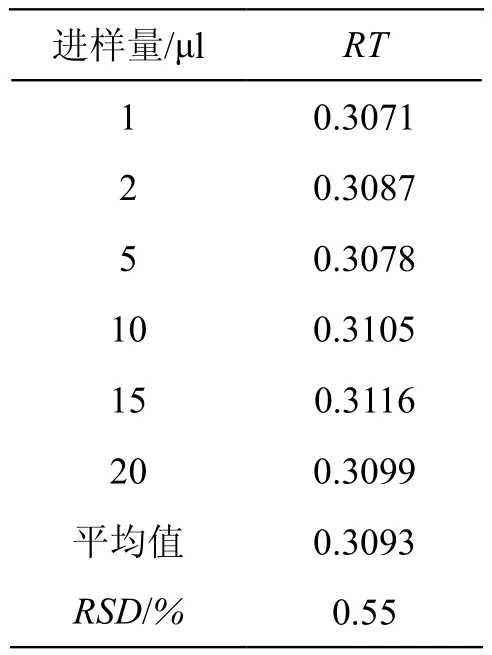
表5 RT值的测定结果
4.3 系统适用性考察
4.3.1 色谱柱对f的影响 采用Agilent 1260高效液相色谱仪,分别考察3种不同品牌的色谱柱对f值的影响,结果见表6。由表6可见,毒黄素与内参物米酵菌酸f值重现性较差(RSD>5 %)。

表6 不同色谱柱对相对校正因子的影响
4.3.2 仪器对f的影响 采用Uitimate®UHPLC AQ-C18(4.6 mm×250 mm,5 μm)色谱柱分别考察3种不同高效液相色谱仪对f的影响结果,结果见表7。由表7可见,毒黄素与内参物米酵菌酸f重现性较差(RSD>5 %)。

表7 不同仪器对f值的影响
4.3.3 流速、柱温、波长对f的影响 采用Agilent 1260高效液相色谱仪和Uitimate®UHPLC AQ-C18(4.6 mm×250 mm,5 μm)色谱柱分别考察3种不同流速(0.9,1.0,1.1 ml/min)、不同柱温(25,30,35 ℃)及不同波长微小变化(波长分别为255,258,260 nm)对f的影响,结果见表8。由表8可见,这3个条件对毒黄素与内参物米酵菌酸f影响较小,重现性良好(RSD<5 %)。

表8 不同色谱条件对f值的影响
4.4 QAMS待测色谱峰的定位
试验中分别考察了米酵菌酸和毒黄素2种成分间RT值在3个不同型号的高效液相色谱仪和3种不同品牌色谱柱中的重现性,见表9。结果显示,RT值变化较大(RSD>5 %),重现性较差。

表9 待测色谱峰的RT值
5 讨论
本试验以湿米粉为例,通过方法学验证、方法的耐用性、系统适用性考察,探讨应用QAMS检测椰毒假单胞菌酵米面亚种两种代谢产物毒黄素和米酵菌酸的可行性。试验中对不同液相色谱仪、不同色谱柱、不同流速、不同柱温和不同波长对f的影响进行考察。其中不同液相色谱仪与不同色谱柱使f变化大,重现性差,RT值不稳定。结果表明,QAMS在实际应用中存在一定的局限性,不适用于毒黄素与米酵菌酸的测定。其原因可能有以下几点。
5.1 化合物类型(母核)不同
据相关研究,QAMS应用于同类型化合物(母核相同、仅仅是取代基的差异)的同时测定是完全没有问题的,而对于不同类型的化合物,现有研究结果尚无法得出确定性的结论[9]。不同类型化合物的基本母核不一样,导致其紫外吸收不同。米酵菌酸是一种脂溶性酸性化合物,分子式 C28H38O7,相对分子质量486.61, 化学名为3-羧甲基-17-甲氧基-6, 8, 21-三甲基二十二碳-2, 4, 8, 12, 14, 18, 20-七烯二酸[10],结构见图1。毒黄素为水溶性黄色素,分子式为 C7H7N5O2,相对分子质量193.21 ,化学名1, 6-二甲基-5, 7-二氧-1, 5, 6, 7-四氢嘧啶基(5,4e)非对称三嗪,结构见图2。二者基本母核不同。
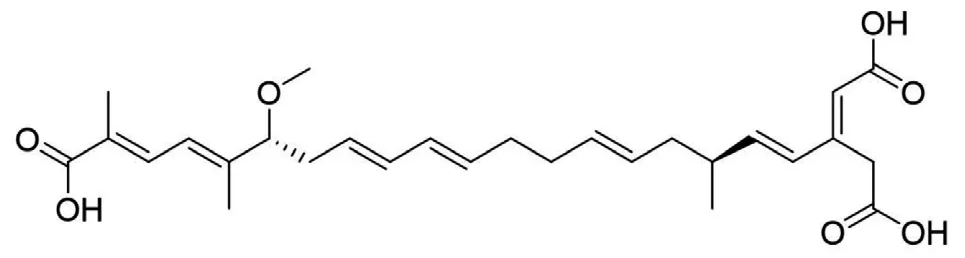
图1 米酵菌酸结构
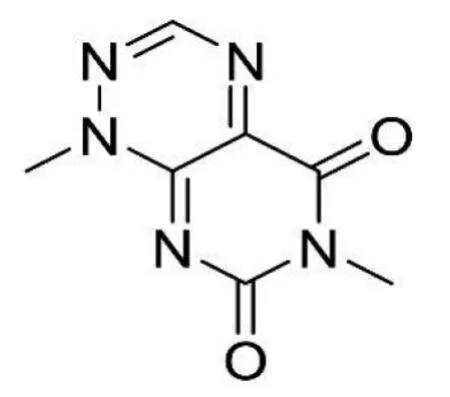
图2 毒黄素结构
研究报道表明,QAMS多应用于中药成分含量研究,食品领域研究报道较少,且多集中在保健食品方面[9]。QAMS应用于同类型化合物的测定被证实是完全可行的,但对于不同类型的化合物,尚有待进一步的研究,其应用具有一定的局限性[11]。
据分析,目前报道的QAMS法,主要针对紫外共轭吸收强的多酚类化合物,使用HPLC/UV或UPLC/UV进行测定,但在食品中还有大量紫外吸收弱、甚至无吸收的化合物[9]。本试验中虽然毒黄素分子结构中含若干共轭键,但米酵菌酸分子多是单键连接,紫外吸收较弱,无法与毒黄素在紫外吸收峰面积上构成正比关系,进而导致其f值的不稳定,在不同系统中存在较大差异。
本研究对GB 5009.189-2016[12]条件进行优化,所建方法可有效解决样品中杂质干扰的问题,并可实现毒黄素和米酵菌酸的有效分离。在此基础上建立食品中毒黄素和米酵菌酸的QAMS测定方法,但在更换液相色谱仪条件下,f值及RT值重现性较差,难以实现以米酵菌酸为参照物,对食品中毒黄素含量进行同时测定。更优化的方法有待进一步研究探讨。
猜你喜欢
传媒评论(2022年10期)2022-12-26
山西医科大学学报(2021年10期)2021-11-18
食品安全导刊(2021年21期)2021-08-30
天津大学学报(自然科学与工程技术版)(2021年9期)2021-06-01
科学与财富(2021年35期)2021-05-10
四川农业大学学报(2020年6期)2021-01-18
食品安全导刊(2020年14期)2020-12-04
食品安全导刊·中旬刊(2020年5期)2020-06-04
世界科学技术-中医药现代化(2020年10期)2020-04-06
海峡姐妹(2019年8期)2019-09-03
