
结核分枝杆菌的耐药机制研究进展
2021-12-15 07:46刁乃超李健明
中国人兽共患病学报 2021年11期
郑 伟,田 甜,王 琦,刁乃超,李健明,时 坤,杜 锐,3,4
结核病(Tuberculosis,TB)是由结核分枝杆菌(Mycobacteriumtuberculosis,M.tb)通过气溶胶或飞沫进行传播引起的[1],是威胁人类健康的世界三大传染病之一。M.tb主要侵袭宿主的肺部,形成肺结核,同时在脑膜,肠和腹膜等部位也会引起继发感染[2]。研究表明艾滋病(AIDS)和结核病同时感染免疫力低下的群体,会带来更严重的危害如高死亡率[3]。世界卫生组织公布的2020年结核病报告中指出, 2019年全球新发结核病患者约有996万,中国新发结核病患者约有83.3万,中国与印度和俄罗斯联邦同为三大结核病高发国家[4]。为解决结核病给世界带来的健康危害,20世纪七八十年代许多科学家进行了抗结核药物的临床研究。目前常用的一线抗结核药物有异烟肼、利福平和已胺丁醇等,二线抗结核药物有氟诺喹酮类、阿米卡星和卡那霉素等,研发的新药有利奈唑胺、贝达喹啉和德拉马尼等。但因抗结核药物的不规范使用和结核分枝杆菌自身细胞壁结构等特点,导致结核分枝杆菌对抗结核药物产生耐药性[5],给结核病的防治带来了严峻的挑战。
结核分枝杆菌对药物产生耐药性是自身生存的一种本能。其对抗结核病药物的抗性主要是由染色体上的基因或单核苷酸多态性(SNP)突变介导的[6]。抗结核病药物的不规范使用,提高了结核分枝杆菌对正在使用的抗结核病药物发生耐药性的概率。如分枝菌酸中某些基因(如rpoB或gyrA)发生突变,降低了抗结核病药物与靶基因的结合能力,削弱药物治疗效果[7]。结核病的治疗要把握黄金时期,在确诊后的前2个月,服用一线药物(异烟肼、利福平、乙胺丁醇和吡嗪酰胺),之后再服用4个月的异烟肼和利福平[8],提高结核病治疗效果的同时降低了耐药发生的概率。由于目前多药耐药和耐多药结核病的发生概率逐年增加,因此通过分析结核分枝杆菌自身组成成分及其对抗结核病药物产生抗性的分子机制,有利于开发新的药物靶点,提高结核病治愈率。
1 原发性耐药(primary drug resistance)
若宿主未接受过抗结核病药物的治疗,结核分枝杆菌产生耐药性的现象称为原发性耐药。da Silva等[9]发现原发性耐药的出现与结核分枝杆菌组成结构密切相关,当结核分枝杆菌组成结构发生变化,降低其对抗菌药物的敏感性从而引发原发性耐药。与革兰氏阴性菌的细胞壁相比,原发性耐药降低了结核分枝杆菌细胞壁的渗透性,从而干扰营养物质或药物的运输[10],同时低渗透性对宿主体内的分枝杆菌起到保护作用,使结核分枝杆菌逃避宿主的免疫监视[10]。
近年来随着结核分枝杆菌结构不断被解析,Batt等人对其结构进行了深层次的阐述(图1),结核分枝杆菌细胞壁主要由脂质和碳水化合物构成,其中脂质大约占40%[10]。其细胞壁分为内外两层,内层由肽聚糖、阿拉伯半乳糖和霉菌酸共价连接在一起,形成肽聚糖-阿拉伯半乳糖-霉菌酸MA-AG-PG复合物(mAGP),构成其渗透性屏障[11]。外层由脂质和蛋白质组成,其中脂质可与细胞壁自由结合。细胞壁中的肽聚糖是菌体所特有的,是由N-乙酰基胞壁酸和N-乙酰葡萄糖胺通过-1,4糖苷键连接成的糖聚骨架,肽聚糖网状排列方式有利于维持菌体形态,可为菌体的生长繁殖提供足够空间[12]。阿拉伯半乳糖由半乳糖和阿拉伯糖残基组成,是肽聚糖与霉菌酸连接的桥梁。Birch等[13]研究发现Rv2673是M.tb的阿拉伯呋喃糖基转移酶C,可将Araf残基从DPA转移到阿拉伯多糖结构域形成α(1→3)Araf残基导致分枝杆菌AG非还原性末端Ara6基序远端的分支阿拉伯多糖结构域。Rv2673缺失会影响结核分枝杆菌细胞壁中阿拉伯糖结构的形成。Rv2673的过表达促进脂多糖合成,降低宿主细胞对结核分枝杆菌的免疫应答能力。由M.tb的多聚磷酸激酶(PPK)和胞外多聚磷酸酶(PPX)分别参与无机多聚磷酸盐(poly(P))的合成和水解的反应,该反应在M.tb持久性中具有重要的调节作用。多聚(P)的积累与M.tb生长受限有关。Rv1026是一种具有长链多聚磷酸酶水解活性的新型胞外多聚磷酸酶。Shi等[14]发现,M.tb在巨噬细胞感染期间,结核多聚(P)的累积导致生长减慢和对异烟肼的敏感性降低,对热和酸性pH的抵抗力增强,并增强细胞内存活,同时低渗透性对宿主体内的结核分枝杆菌的生长起到保护作用,引起机体发病。细胞壁的渗透性主要由霉菌酸决定,霉菌酸主要由长链α-烷基-β-羟基脂肪酸构成[15],其脂肪酸长度的增加会抑制菌体对亲脂分子的吸收[16]。它以海藻糖单菌酸或海藻糖二菌酸的形式存在于菌体细胞壁,形成不对称的外脂双层内小叶结构[15],此结构提高了结核分枝杆菌发生原发性耐药的概率。

图1 结核分枝杆菌细胞壁组成示意图[16]Fig.1 Schematic diagram of the cell wall composition of Mycobacterium tuberculosis[16]
2 获得性耐药(acquired drug resistance)
当宿主的免疫防御机制无法清除入侵的结核分枝杆菌时,就会引发活动性结核病,此时会采用药物进行抗结核病治疗。但当结核分枝杆菌发生基因突变或获得外源性耐药基因时,就会降低结核分枝杆菌对药物的敏感性,发生获得性耐药[17],其中一线药物获得性耐药机制图改编自Khawbung的一线药物作用方式图(图2)[18],二线药物获得性耐药机制改编自Hamdde的与抗结核药物有关的蛋白质/RNA/基因图(图3)[19]。耐药结核病可分为单耐药(monoresistance-tuberculosis, MR-TB)、多耐药(poly-drug resistant tuberculosis, PDR-TB)、耐多药(multidrug-resistance tuberculosis, MDR-TB)
和广泛耐药结核病(extensive drug resistangce-tuberculosis, XDR-TB)。对利福平和异烟肼都耐药的称耐多药结核病;除对一线药物异烟肼和利福平耐药外,还对氟喹诺酮类药物及二线的3种注射类药物(阿米卡星、卡那霉素和卷曲霉素)中至少一种耐药的称为广泛耐药结核病。基因突变是一线药物(如乙胺丁醇)、二线药物(如乙硫异酰胺)和新药(如SQ-109)发生获得性耐药的主要原因。
2.1 一线抗结核病药物
2.1.1 利福平 利福平是对代谢活跃或缓慢的结核分枝杆菌均有活性的药物,它通过与RNA聚合酶β亚基的结合,抑制蛋白质的合成[20],因此结核分枝杆菌中rpoB基因突变是结核分枝杆菌对利福平产生耐药的重要原因。此外,Miotto[21]团队发现利福布汀与利福平存在交叉耐药,当Asp516Ala和Arg529Gln发生双突变,会促进菌体对二者的耐药[22]。Sinha[23]发现了新的耐利福平突变位点:包括528密码子Arg-Cys突变、518密码子Asn-Asp突变、506密码子Phe-Leu突变、511密码子Leu-Pro突变、513密码子Gln-Val/Glu/Pro/Leu突变和510密码子Gln-His突变,新的突变位点的出现是M.tb适应药物暴露的结果。
2.1.2 异烟肼 异烟肼是在1952年引入的抗结核病关键药物,是联合治疗活动性肺结核最有效的药物之一,也是治疗潜伏性肺结核的单一药物。它由过氧化氢酶-过氧化物酶激活,最终形成异烟酰乙酸—NADH复合物[24],可抑制结核分枝杆菌的增殖。katG是编码过氧化氢酶—过氧化物酶的主要基因,当katG基因突变可促进分枝菌酸细胞壁的合成使菌体对异烟肼的耐药[25]。KatG表达与INH-MIC变化呈负相关,当KatG表达减少2倍导致MIC增加略大于2倍。当结核分枝杆菌inhA基因位点发生碱基插入、缺失或突变,是结核分枝杆菌对异烟肼耐药的另一个原因。当结核分枝杆菌对异烟肼和利福平都产生抗性,会引起耐多药结核病。DanA与M.tbRv0010c-Rv0011c基因间区域特异性结合,二者的突变均可介导INH耐药性。DanA突变与高水平耐药有关,danA/Rv0010c-Rv0011c突变产生了对INH中水平耐药[26]。M.tb的Rv2170具有N-乙酰转移酶结构域,能乙酰化INH,是M.tb抗异烟肼的另一新机制[27]。
2.1.3 乙胺丁醇 乙胺丁醇它是一种抗菌药物。它通过抑制阿拉伯糖基转移酶(主要由embCAB操纵子编码)阻断阿拉伯半乳糖的合成,降低细胞壁通透性杀死结核分枝杆菌[28]。研究发现结核分枝杆菌中ubiA和embB基因突变会提高结核杆菌对乙胺丁醇的耐药性,但具体机理还需加以验证[29]。EmbR转录因子调控embCAB操纵子,embR的磷酸化可调控其活性,促进其与DNA结合,导致embCAB基因转录增加,抗性改变[30]。
2.1.4 吡嗪酰胺 吡嗪酰胺与乙胺丁醇都是治疗耐多药结核病的抗菌药物,吡嗪酰胺由pncA编码的吡嗪酰胺酶水解为吡嗪酸。当pncA基因发生突变,吡嗪酸会抑制菌体细胞膜的功能,干扰细菌膜与胞内胞外的物质转运,使菌体对吡嗪酰胺产生耐药性。研究表明除pncA外,rpsA和panD突变也会使菌体产生耐药性[31]。rpsA突变后与吡嗪酸相互作用,促进tmRNA的传递,加速结核分枝杆菌“反式翻译”(trans-translation)过程[17]。panD发生突变有助于吡嗪酸加速辅酶A与泛酸盐的反应,促进结核分枝杆菌新陈代谢,减弱药物抗菌作用使菌体产生耐药性。Zhang等[32]对吡嗪酰胺进行了药效与毒力学分析,发现吡嗪酰胺的药效与其浓度成正比,但吡嗪酰胺必须联合利福平同时使用才能缩短结核病的治疗时间。
2.1.5 链霉素 链霉素是从灰链霉菌培养液中提取的。它通过与结核分枝杆菌核糖体30S亚基16S rRNA和S12核糖体蛋白作用,阻碍tRNA与30S亚基的结合,干扰蛋白质的生物合成[33],造成菌体裂解死亡。因此编码核糖体30S亚基16S rRNA的rrs基因和编码S12核糖体蛋白的rpsL基因突变是结核分枝杆菌对链霉素耐药的主要原因。

注:利福平靶点位于RNA聚合酶β亚基;异烟肼靶点为InhA和KatG基因;乙胺丁醇靶点为Emb-B基因;吡嗪酰胺靶点为PncA,RpsA和PanD基因;链霉素靶点在Rrs和RpsL基因中,当药物作用的靶点基因发生突变会引发结核分枝杆菌对一线药物的耐药性。图2 一线药物获得性耐药机制图Fig.2 Diagram of the mechanism of first-line drug-acquired resistance
2.2 二线抗结核病药物
2.2.1 阿米卡星、卡那霉素、卷曲霉素 当结核病患者对一线抗结核病药物产生耐药性,临床上会联合二线抗结核病药物继续进行治疗。其中二线抗结核病注射类药物阿米卡星和卡那霉素通过与菌体16S核糖体RNA结合,抑制菌体蛋白的合成[34]。编码16S核糖体RNA的rrs基因突变,会削弱阿米卡星与卡那霉素对结核分枝杆菌的抑制作用。Islam团队[35]发现,编码氨基糖苷乙酰基转移酶的eis基因突变,结核分枝杆菌会对阿米卡星和卡那霉素产生低水平的抗性。卷曲霉素是1979年首次用于治疗结核病的多肽类抗生素,研究发现,临床上单用卷曲霉素治疗结核病极易产生耐药性[36],必须联合其它抗结核病药物(如异烟肼和乙胺丁醇)一起使用。
2.2.2 乙硫异酰胺 临床上治疗抗结核病的二线药物除注射剂外还有乙硫异酰胺。乙硫异酰胺是一种治疗结核病的前药,其抗菌活性远低于异烟肼。编码黄素单加氧酶的ethA或NADH特异性烯酰基载体蛋白还原酶的inhA基因发生突变[37],有助于细胞壁中霉菌酸的合成,降低其通透性。Mugumbate[38]团队发现ethR基因过表达,降低ethA蛋白活性,延缓转录,进而减弱了乙硫异酰胺对结核分枝杆菌的抑制作用。通过溶解曲线评估inhA突变是否可作为预测乙硫异酰胺耐药性的分子标志,因突变的inhA可能不对乙硫异酰胺产生抗性,所以一般不用inhA基因检测该药的耐药性[39]。
2.2.3 氟喹诺酮类 氟喹诺酮类是治疗广泛耐药结核病的首选药物。研究发现,DNA促旋酶是氟喹诺酮类药物作用的靶标,编码DNA促旋酶的gyrA和gyrB基因发生突变,有利于菌体DNA的合成,加速结核分枝杆菌的分裂,使菌体产生耐药性[40]。Chawla[41]团队在印度南部某三级医疗中心进行结核分枝杆菌对氟喹诺酮耐药基因的检测,发现gyrA在第90、91和94位密码子和gyrB基因G1498A的突变较为常见。
2.2.4 对氨基水杨酸 对氨基水杨酸虽属于对氨基苯甲酸同似物,但其在治疗疾病时与对氨基苯甲酸起拮抗作用,是治疗结核病的抑菌剂。其通过调节叶酸的合成[42],控制菌体代谢。当结核分枝杆菌Folc基因发生突变,会加速菌体内叶酸的合成速度,提高菌体对该药代谢速率产生耐药性[43]。异烟肼或乙胺丁醇等一线抗结核病药物与对氨基水杨酸联合使用可延缓结核分枝杆菌耐药性的发生。Wang[42]团队发现丙烯胺N-乙酰基转移酶过表达,对氨基水杨酸的最小抑菌浓度将提高2倍,减弱宿主内对氨基水杨酸对分枝菌酸的抗菌作用。
2.2.5 D-环丝氨酸 D-环丝氨酸是抑制结核分枝杆菌活性弱于链霉素的二线抗结核病药物,该药是治疗耐多药与广泛耐药结核病的常用药物。它可抑制菌体细胞壁肽聚糖合成关键酶:D-丙氨酸消旋酶和D-丙氨酸连接酶,阻止肽聚糖的生物合成,降低细胞壁渗透性[44],促进菌体与药物结合,消灭宿主体内的结核分枝杆菌。因细菌对其产生耐药性的几率较小,所以此药常用于受结核分枝杆菌侵染人群的治疗。研究表明,结核分枝杆菌alr、ald、ddlA和cycA基因发生突变会对D-环丝氨酸产生耐药性[45]。这些基因发生突变,增加菌体细胞壁的厚度,使药物难以渗透过细胞壁,令菌体对D-环丝氨酸产生耐药性。
2.2.6 利奈唑胺 利奈唑胺是人工合成的恶唑烷酮类抗生素,它通过与50S核糖体亚基上肽基转移酶中心的tRNA的结合与抑制,阻碍结核分枝杆菌生长,其在临床上与其他抗菌药较少发生交叉耐药现象,且在体外也不易诱导细菌发生耐药[46]。Bolhuis等[47]研究发现,利奈唑胺可破坏结核分枝杆菌细胞壁完整性,令其它药物(如异烟肼)更易于通过,达到杀死结核分枝杆菌的目的。当结核分枝杆菌rrl和rplc基因发生突变时,有助于促进菌体蛋白的合成,为结核分枝杆菌的繁殖提供足够的营养,降低利奈唑胺对结核分枝杆菌活性的抑制作用。Fermeli[48]团队发现长期服用该药会导致严重的不良反应,如血小板减少、乳酸中毒和眼部神经病变等。
2.2.7 氯法齐明 氯法齐明是治疗麻风病和结核病的抗真菌药物,Gopal[49]团队发现当巨噬细胞中含有高浓度的氯法齐明,其会抑制结核分枝杆菌衍生因子对吞噬细胞的杀伤作用,抑制菌体生长繁殖。氯法齐明通过干扰菌体蛋白的合成,抑制结核分枝杆菌的活性。Mothiba等人通过肉汤和平板稀释法发现,当氯法齐明的MIC为0.06 mg/L可抑制结核分枝杆菌的生长,当MIC为2.5 mg/L时可抑制耻垢分枝杆菌的生长[50]。研究发现,结核分枝杆菌Rv0678基因的非靶向突变会使外排泵发生上调,进而加快菌体生长速度,使分枝菌酸对氯法齐明发生耐药[22]。氯法齐明可促进中央记忆T细胞免疫应答,通过阻断Kv1.3的T细胞表面上钾离子通道降低效应记忆T细胞群功能,缩短耐多药结核病的治疗时间[51]。
2.3 新 药
2.3.1 贝达喹啉 贝达喹啉多用于耐多药结核病的治疗,其可消灭宿主体内潜伏的结核分枝杆菌。它通过与ATP合酶的α和c亚基紧密结合(由atpE基因编码)调节结核分枝杆菌活性,当atpE基因发生突变,削弱了贝达喹啉抑菌或杀菌作用,令结核分枝杆菌对该药产生抗性。因贝达喹啉消除半衰期需5~6个月,明显长于其他抗结核病药物,所以在临床用药时应考虑其药代动力学特征[52]。Ismail等[53]发现结核分枝杆菌中编码MmpR5阻遏蛋白的Rv0678发生突变,将会促进MmpL5-MmpS5外排泵的过表达,减弱药物与靶点的结合能力。Ismail[54]在另一项研究中发现atpE突变出现的频率与Rv0678突变频率成正比,且在低MIC条件下有利于Rv0678基因发生突变,反之则利于atpE基因发生突变。
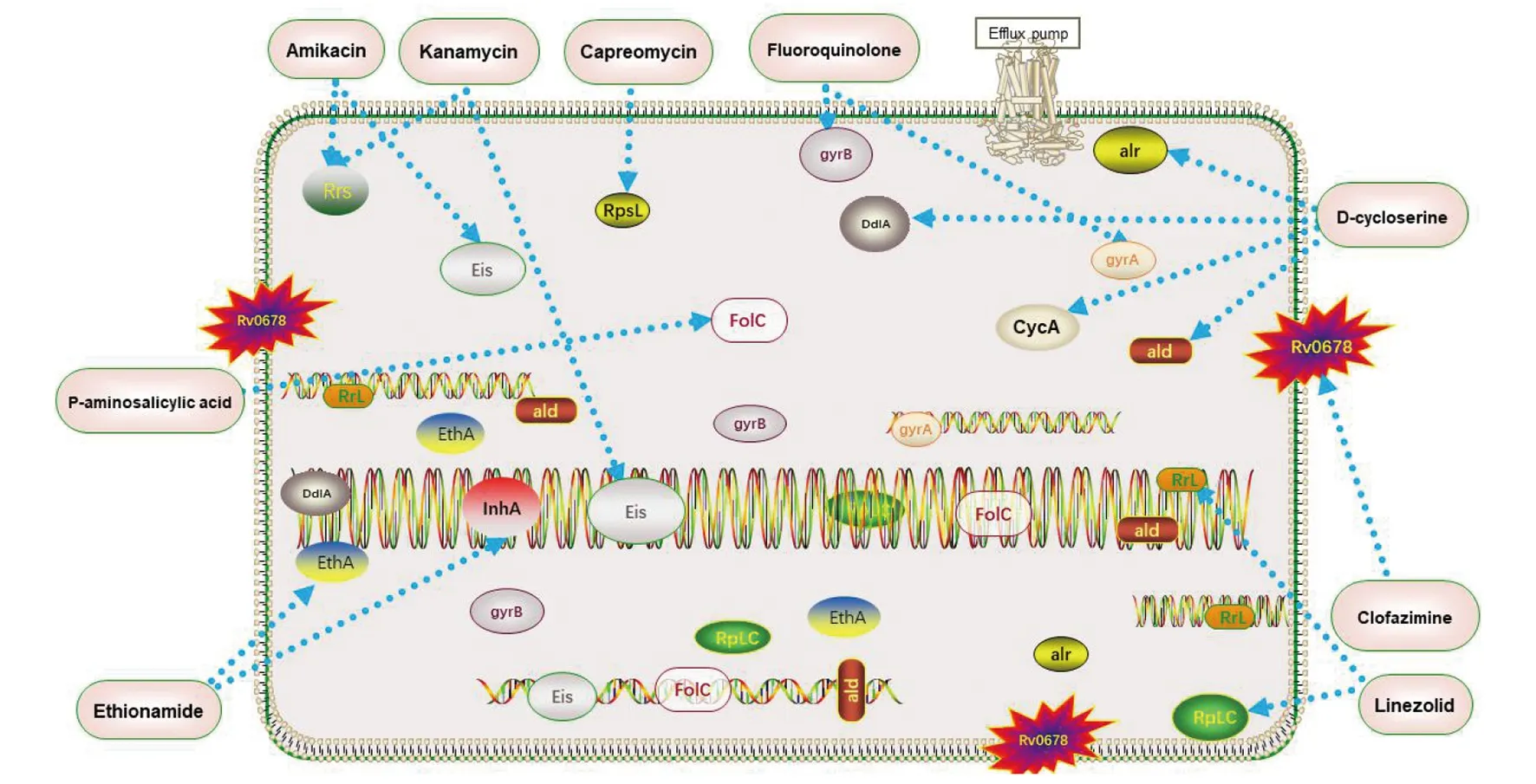
注:阿米卡星和卡那霉素都作用于Rrs和Eis基因;卷曲霉素作用于RpsL基因;乙硫异酰胺作用于EthA和InhA基因;氟喹诺酮类药物作用于gyrA和gyrB基因;对氨基水杨酸作用于FolC基因;D-环丝氨酸作用于alr,DdlA和CycA基因;利奈唑胺作用于RrL和RplC基因;氯法齐明作用于Rv0678,调控细胞壁渗透性,当药物作用的靶点基因发生突变会引发结核分枝杆菌对二线药物的耐药性。图3 二线药物获得性耐药机制图Fig.3 Diagram of acquired resistance mechanism of second-line drugs
2.3.2 德拉马尼和Pretomanid 德拉马尼与Pretomanid都是治疗结核病的新药,它们的出现给结核病患者带来新的福音。德拉马尼是一种新型分枝杆菌细胞壁合成抑制剂,它通过阻碍细胞壁霉菌酸的合成,提高细胞壁的渗透性,达到杀死结核分枝杆菌的目的[55],反之当结核分枝杆菌中ddn、fgd1和fbiA/B/C基因发生突变则会令菌体对德拉马尼产生耐药性。Mallikaarjun[56]发现当德拉马尼与利福平和乙胺丁醇同时用于结核病患者治疗时,宿主会减少对德拉马尼的吸收。Liu[57]的团队根据临床数据推测因德拉马尼有良好的耐受性,所以可能不易产生致癌性。Pretomanid属于硝基咪唑类抗生素,是抗结核病的前药,它对潜在或耐多药结核分枝杆菌均有抑菌效果。当结核分枝杆菌进行繁殖时,Pretomanid抑制细胞壁霉菌酸的合成,进而抑制结核分枝杆菌的增殖,在非复制条件下,Pretomanid被脱氮黄素依赖的硝基还原酶降解,产生具有较强抗真菌活性的氮,阻止结核分枝杆菌的繁殖[58]。研究发现当结核分枝杆菌中Rv3547和Rv0407基因发生突变时,破坏细胞壁的完整性,降低Pretomanid抑菌作用,令结核分枝杆菌对Pretomanid产生抗性[22]。
2.3.3 SQ-109 SQ-109对广泛耐药结核病有较好的治疗效果,是一种不对称二胺,其对结核分枝杆菌的抑制作用与MmpL3基因密切相关[59]。MmpL3是将霉菌酸运输到结核分枝杆菌外膜所必须的基因,当MmpL3基因发生突变,将加速霉菌酸的合成,降低细胞壁通透性,减弱SQ-109对结核分枝杆菌的杀伤作用,令菌体产生耐药性[60]。当SQ-109与利福平、贝达喹啉和异烟肼共同使用时,SQ-109与利福平的协同作用更显著。Jia采用SQ-109、异烟肼与乙胺丁醇共同治疗已被结核分枝杆菌侵染的小鼠,检测SQ-109的抗菌活性,得出结论:SQ-109抑菌活性与异烟肼相似,且更优于乙胺丁醇[61]。
3 展 望
由于抗结核病药物的不规范使用,结核分枝杆菌的不断进化,使其可以逃避宿主的免疫监控,降低了抗结核病药物对结核分枝杆菌的抑制作用。研究结核分枝杆菌的耐药机制,寻找新的药物靶点,开发最新型抗结核病药物已迫在眉睫。通过分析结核分枝杆菌发生耐药性的原因,其一可避免在治疗结核病时使用已发生耐药性的药物,提高结核病治愈的几率;其二有助于发现新型药物靶点,促进对新型抗结核病药物研发。新型抗结核病药物的研发将大大提高结核病患者的康复率,所以我们必须重视结核分枝杆菌的耐药机制。
利益冲突:无
引用本文格式:郑伟,田甜,王琦,等.结核分枝杆菌的耐药机制研究进展[J].中国人兽共患病学报,2021,37(11):1044-1052. DOI:10.3969/j.issn.1002-2694.2021.00.148
猜你喜欢
化工时刊(2022年4期)2023-01-06
山东化工(2020年20期)2020-11-25
三农资讯半月报(2020年18期)2020-10-14
数码世界(2018年1期)2018-12-23
浙江工业大学学报(2017年5期)2018-01-22
广东农业科学(2017年5期)2017-08-29
现代检验医学杂志(2016年2期)2016-11-14
中国继续医学教育(2015年1期)2016-01-06
中外医疗(2015年11期)2016-01-04
中国卫生标准管理(2015年13期)2015-01-26
