
基于公共数据库的甲状腺癌预后风险基因筛选
2021-12-15 08:06:54曾杰杨秋怡张志鹏范培芝张超杰廖雯
中国普通外科杂志 2021年11期
曾杰,杨秋怡,张志鹏,范培芝,张超杰,廖雯
(1.湖南师范大学附属第一医院/湖南省人民医院乳甲外科,湖南长沙410005;2.中南大学湘雅医院老年外科,湖南长沙410008)
甲状腺癌是最常见的内分泌系统的恶性肿瘤,其发病率在过去10年间持续攀升[1]。它的平均年增长率约为6.6%,在2000—2009年美国所有癌症中增长速度最高[2]。在过去几十年里,甲状腺癌在许多国家达到了3 倍的增长率[3-4]。最新的全球癌症统计资料显示,2018年全球甲状腺癌的新发病例为567 000 例,在发病率中排名第九,占所有癌症新发病例的5.1%,且女性发病率是男性的3 倍多[5]。值得庆幸的是,其病死率较低,约占所有癌症死亡病例的0.4%~0.5%[5]。甲状腺癌起源于滤泡上皮细胞或滤泡旁C 细胞,其中滤泡上皮来源的甲状腺癌可分为四种病理类型:乳头状癌(80%~85%)、滤泡状癌(10%~15%)、低分化癌(<2%)和未分化癌(<2%),乳头状癌和滤泡状癌合称为高分化甲状腺癌,占全部甲状腺癌的绝大多数[6]。尽管通过手术、甲状腺激素和放射性碘治疗后,高分化甲状腺癌的预后较好,但仍有一部分患者因复发或转移而死亡[6-7]。此外,甲状腺细针穿刺细胞学检查是甲状腺癌术前诊断最可靠的检查手段,但该检查结果中仍然有10%~40%具有不确定性[8-9]。而且,单纯依靠细针穿刺细胞学检查诊断甲状腺微小乳头状癌也有较高的假阴性率[10]。有研究[11]报道,细针穿刺细胞学检查联合生物标志物检测可提高甲状腺癌的诊断准确率。因此,发现和探索新的生物标志物或治疗靶点可能有助于改善甲状腺癌患者的诊断和治疗效果。
1 资料与方法
1.1 数据来源
本研究从癌症RNA 测序关系(Cancer RNA-seq Nexus,CRN)数据库下载甲状腺癌的蛋白编码基因RNA-seq 数据[12],总计564 例样本,其中癌旁正常组织59 例,甲状腺癌组织505 例,后者又再分为I 期283 例、II 期53 例、III 期112 例,IVA 期46 例、IVC 期6 例和MI 期5 例。
1.2 筛选甲状腺癌中差异表达的蛋白编码基因
由于CRN 数据库是从癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库下载的甲状腺癌的蛋白编码基因RNA-seq 数据,并对这些数据进行了注释、标准化及差异表达分析,将P<0.01 作为统计学差异指标。因此,我们可以直接使用从CRN 数据库中筛选出来的蛋白编码基因进行后续研究。为了保证数据的可靠性,取甲状腺癌各临床分期中差异表达蛋白编码基因的交集进行研究,通过omicshare(http://www.omicshare.com)云平台绘制Venn 图[13]。
1.3 差异表达蛋白编码基因的功能预测
DAVID(https://david.ncifcrf.gov)数据库为研究人员提供了一套全面的功能注释工具,以理解大量基因背后的生物学意义[14]。通过DAVID 数据库对上述得到的差异表达蛋白编码基因进行功能富集分析,分析内容包括基因本体论(gene ontology,GO)的生物学过程(biological process,BP)、细胞成分 (cellular component, CC)、 分子功能(molecular function,MF)以及京都基因和基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG) 的信号通路。分析结果使用R 语言的ggplot2 包呈现出来,P<0.05 认为有统计学差异。
1.4 筛选差异表达蛋白编码基因中的核心基因
STRING(https://www.string-db.org/)是一个分析已知或预测蛋白质之间相互作用的数据库[15]。通过STRING 数据库对上述筛选到的差异表达蛋白编码基因之间的相互作用网络进行预测和可视化,将有实验证实且互作分数>0.4 的蛋白编码基因筛选出来,并在Cytoscape 软件中重建互作网络。使用Cytoscape 软件中cytoHubba 插件筛选核心基因,使用ClueGO 插件对核心基因进行功能预测[16-18]。
1.5 验证核心基因在甲状腺癌中的表达水平
UALCAN (http://ualcan.path.uab.edu) 数据库为用户提供了简易获取肿瘤OMICS 数据(包括TCGA、MET500 和CPTAC 数据资源)的途径,允许用户对感兴趣的基因进行生物信息学验证,并可以对基因的表达水平进行图像展示[19]。本研究通过UALCAN 数据库验证核心基因在甲状腺癌中的表达水平,P<0.05 认为有统计学意义。
1.6 核心基因在甲状腺癌中的生存分析
GEPIA (http://gepia.cancer-pku.cn) 是一个新开发的用于分析TCGA 和GTEx 项目收录的癌症RNAseq 数据的网站,它可为用户提供恶性肿瘤的差异表达分析、生存分析、相似基因检测、相关性分析和降维分析[20]。本研究基于GEPIA 数据库分析核心基因对甲状腺癌患者生存状态的影响,P<0.05 认为差异有统计学意义。
2 结果
2.1 筛选甲状腺癌中差异表达的蛋白编码基因
通过比较各临床分期甲状腺癌与癌旁正常组织中蛋白编码基因的表达水平,得到各临床分期中差异表达的蛋白编码基因,再取各临床分期中差异表达蛋白编码基因的交集进行后续研究。最终,本研究共筛选得到913 个差异表达的蛋白编码基因(图1)。
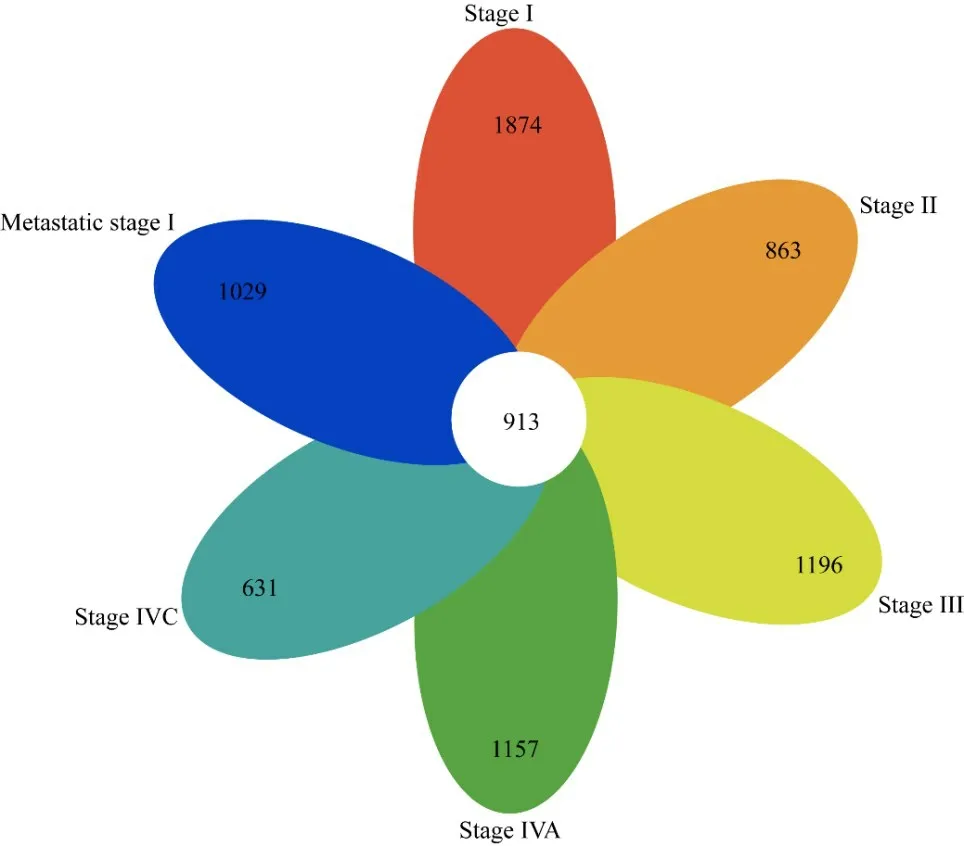
图1 甲状腺癌各临床分期中差异表达蛋白编码基因的Venn图(不同颜色花瓣中的数字代表各临床分期中差异表达蛋白编码基因的数目,图片中央白色区域中的数字为各临床分期中差异表达蛋白编码基因的交集)Figure 1 Venn diagram of the differentially expressed protein-coding genes in thyroid cancer of different clinical stages(the number in petals with different colors representing the number of the differentially expressed protein-coding genes of different clinical stages,and the number in the central region in white color representing the overlap of the differentially expressed protein-coding genes of different clinical stages)
2.2 差异表达蛋白编码基因的功能预测
借助于DAVID 数据库,本研究对上述得到的913 个差异表达的蛋白编码基因进行功能富集分析,每个项目分析后的前10 个结果以气泡图的形式展现出来(图2)。结果显示,上述得到的差异表达蛋白编码基因可能参与了调控小分子GTP 酶介导的信号转导(GO-BP)、Z 膜(GO-CC)、结合肌动蛋白(GO-MF)和细胞色素P450 介导的药物代谢(KEGG pathway)。
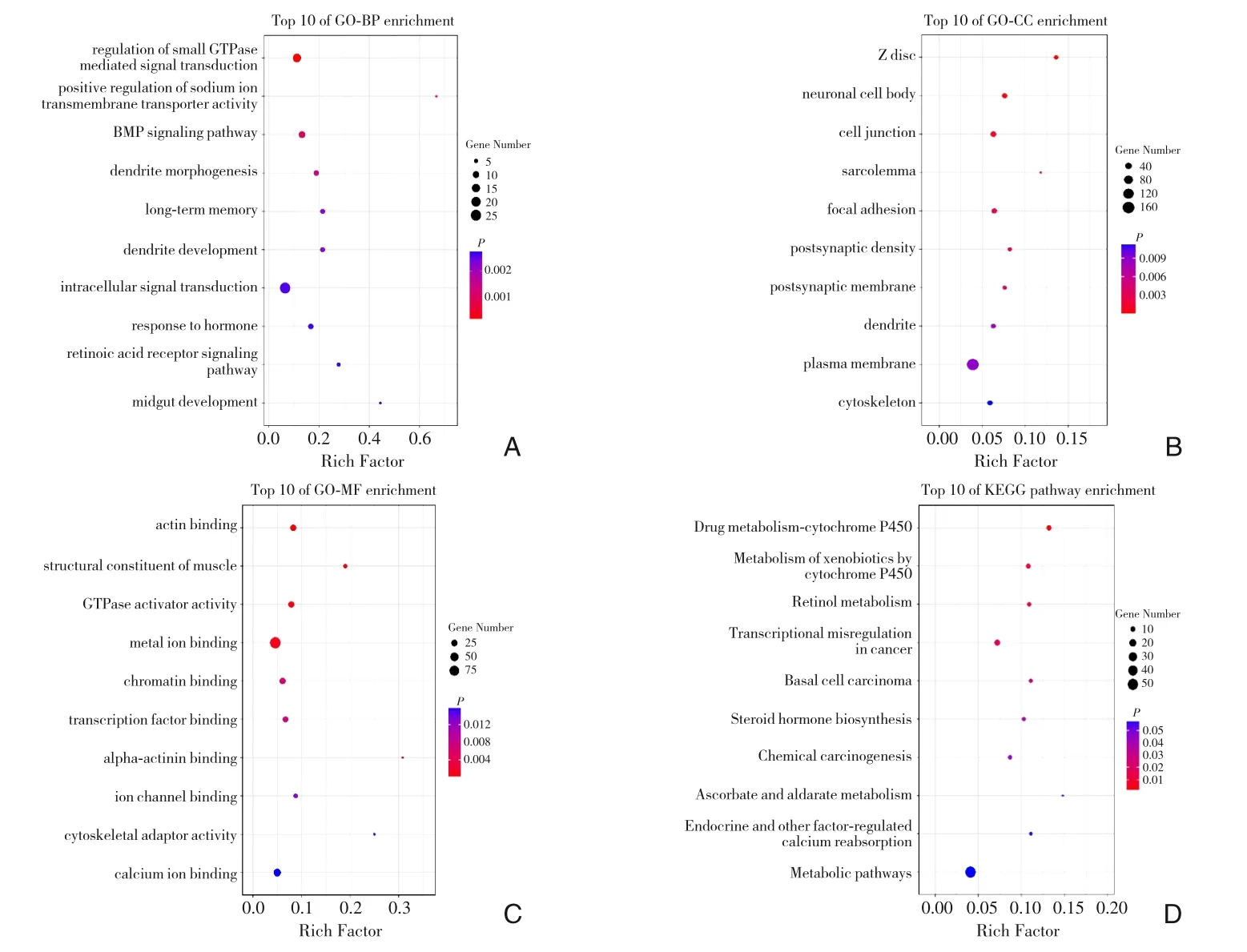
图2 差异表达蛋白编码基因的功能富集分析(左侧的Y 轴显示功能富集分析结果;下方的X 轴表示参与BP、CC、MF 和KEGG的基因所占的百分比;气泡大小表示参与BP、CC、MF和KEGG的基因数目,气泡越大表示参与的基因越多;气泡颜色代表P 值的大小,颜色由红至蓝代表P 值越大)A-D:分别显示了这些差异表达基因可能参与的前10 个BP、CC、MF和KEGG结果Figure 2 Enrichment analysis of the differentially expressed protein-coding genes(the left Y axis showing the results of enrichment analysis,the inferior X axis showing the percentages of genes involved in BP,CC,MF and KEGG,the bigger of the bubble,the larger number of genes involved;the color of the bubble standing for the level of the P-value,and the level increasing from the red color to blue color)A-D:The results of BP,CC,MF and KEGG of the top ten differentially expressed genes
2.3 核心基因的筛选及功能预测
通过Cytoscape 软件重建差异表达蛋白编码基因之间的互作网络, 使用Cytoscape 软件中cytoHubba 插件计算每个节点的得分,将得分最高的前10 个基因定义为核心基因,分别为TP53、ESR1、 FOS、 SYP、 PPARG、 ACTB、 GRIA1、NRXN1、HDAC3 和KIT(图3),其中TP53 得分最高,为62;使用ClueGO 插件对核心基因参与的生物学过程进行功能预测,P<0.05 认为差异有统计学意义,结果显示核心基因TP53、ESR1、PPARG可能参与了基因沉默的负性调控,TP53、FOS 可能参与了RNA 聚合酶II 对pri-miRNA 的转录过程(图4)。

图3 差异表达蛋白编码基因的相互作用网络图(图中的节点代表每个差异表达的蛋白编码基因,黄边粉红色节点代表核心基因)Figure 3 Interaction networks of the differentially expressed protein-coding genes(each node indicating a differentially expressed protein-coding gene,and the nodes in pink color with yellow border indicating the hub genes)
2.4 验证核心基因在甲状腺癌中的表达
从CRN 数据库中下载的数据显示,与癌旁正常组织相比,10 个核心基因在甲状腺癌组织中均呈低表达。本研究再通过UALCAN 数据库验证这10 个核心基因在甲状腺癌组织中的表达水平,结果显示除了TP53 在甲状腺癌组织中呈高表达外,其余核心基因均在甲状腺癌组织中表达下调,差异有统计学意义(图5),与从CRN 数据库中下载的数据资料保持一致。
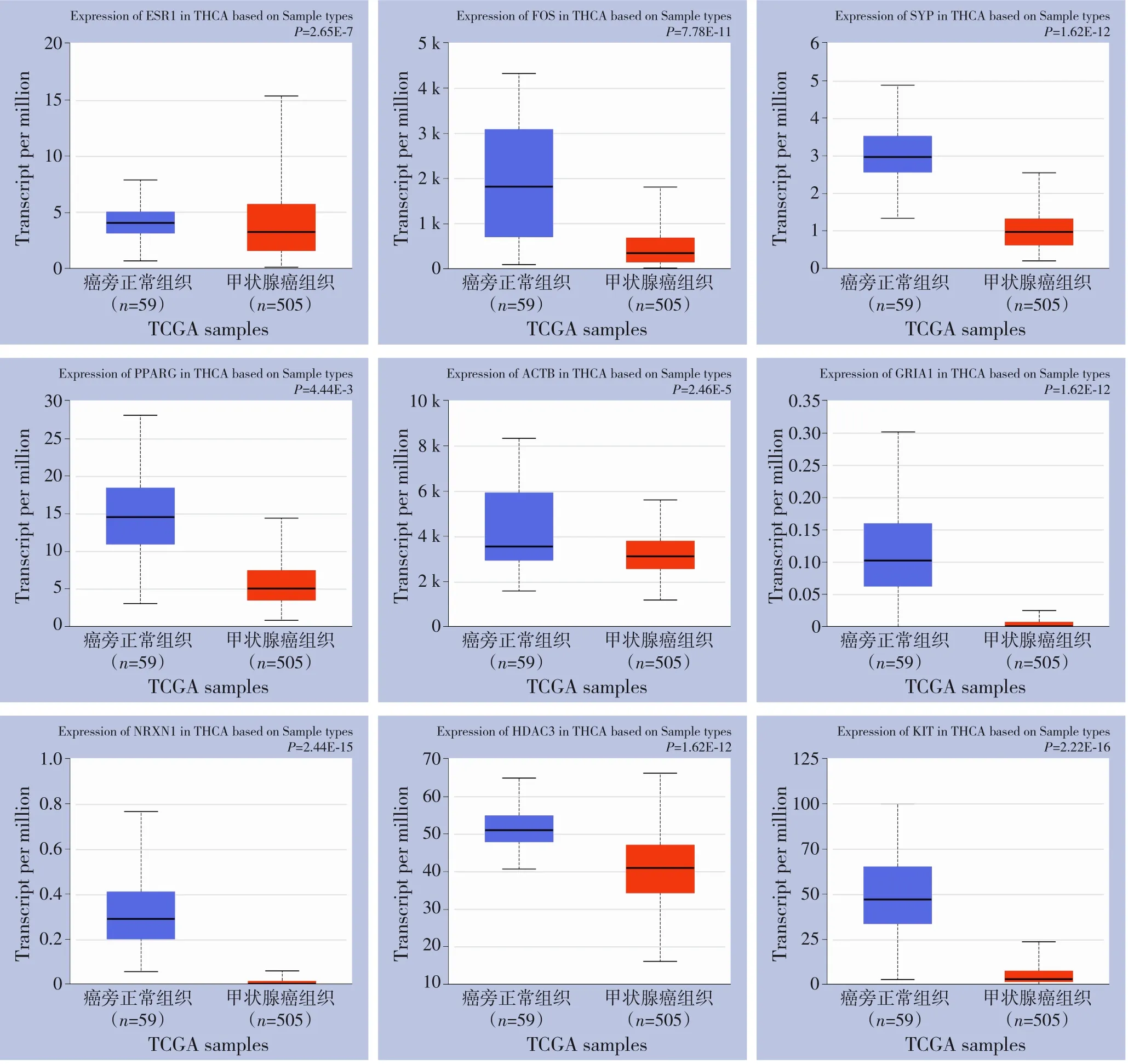
图5 基于UALCAN数据库验证核心基因在甲状腺癌组织与癌旁正常组织中的表达水平(蓝色箱型图代表癌旁正常组织,红色箱型图代表甲状腺癌组织)Figure 5 Verification of the hub genes in thyroid tissue and normal adjacent tissue based on the UALCAN database(the blue box diagram representing the normal paracancer tissue and the red box diagram representing the thyroid cancer tissne)
2.5 核心基因在甲状腺癌中的生存分析
基于GEPIA 数据库中甲状腺癌的生存数据,本研究对这10 个核心基因进行了生存分析,以基因表达的均值作为截断值分为高表达组和低表达组,分析这10 个核心基因对甲状腺癌患者无病生存期及总生存期的影响(部分基因因为样本量太小无法进行生存分析),结果显示与KIT 低表达组相比,KIT 高表达组患者的无病生存期明显延长(P=0.012),但对总体生存期无影响(P=0.85),这可能与甲状腺癌患者的整体预后较好有关(图6)。
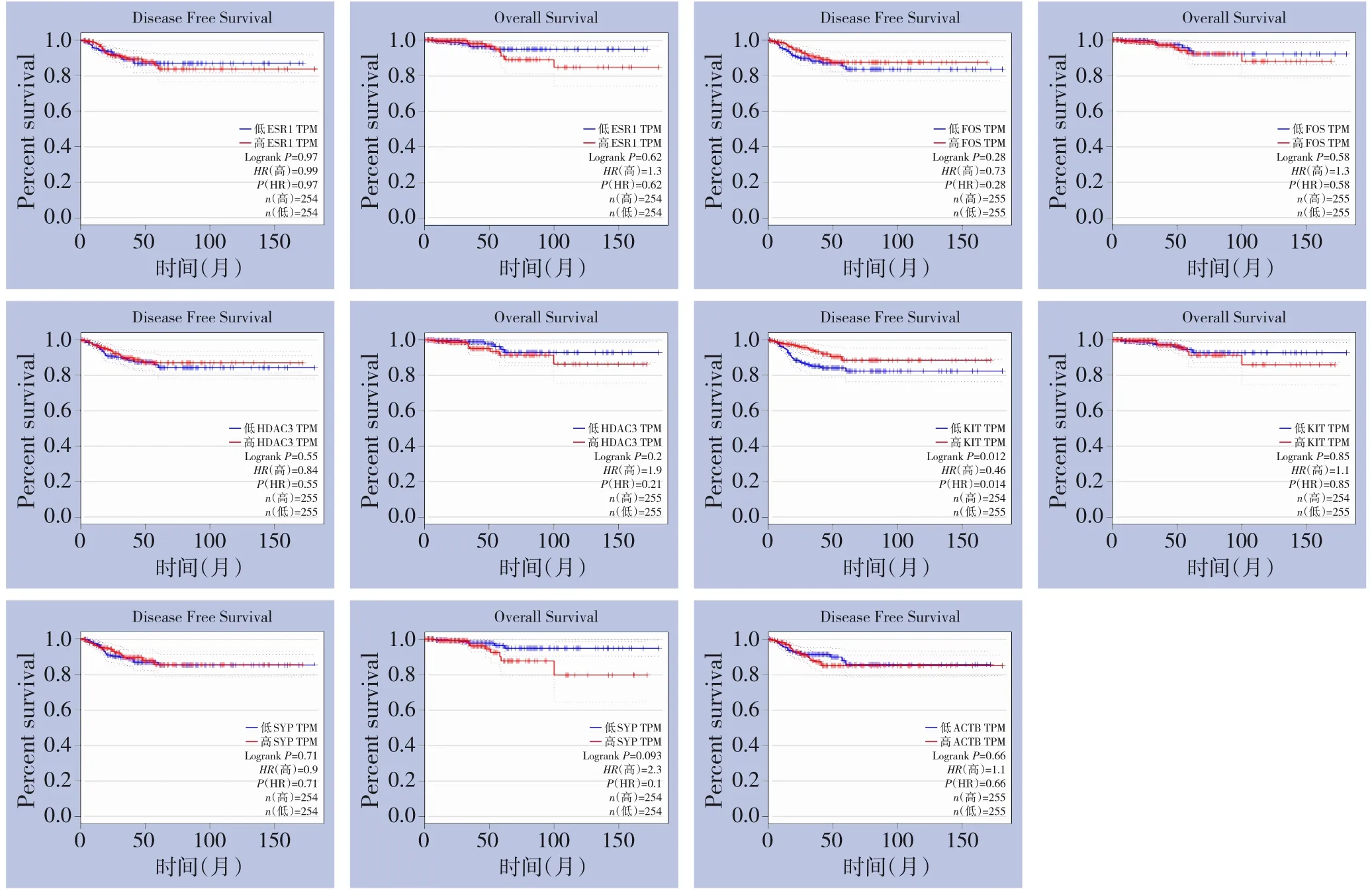
图6 核心基因的生存曲线(红色曲线代表高表达组,蓝色曲线代表低表达组)Figure 6 survival curves of the hub genes(the red curve representing the high-expression group and the blue curve representing the low-expression group)
3 讨论
本研究基于公共数据库通过生物信息学方法筛选出KIT 基因作为甲状腺癌的核心基因,CRN 数据库和UALCAN 数据库证实了KIT 在甲状腺癌组织中呈低表达,GEPIA 数据库中甲状腺癌的生存资料显示KIT 的高表达与甲状腺癌患者的无病生存期明显相关,推测KIT 基因可作为甲状腺癌的预后风险基因或治疗靶点。
KIT,也称为c-KIT,编码酪氨酸激酶受体(CD117),后者是III 型酪氨酸激酶受体家族成员之一。KIT 可通过丝裂原活化蛋白激酶(MAPK)、磷脂酰肌醇3-激酶(PI3K),Janus 激酶(JAK)/信号转导和转录活化因子(STAT),SRC 家族激酶(SFK)和磷脂酶Cγ 等多条下游信号通路参与细胞增殖、凋亡、化学趋向和黏附[21-22]。此外,KIT 是一种诱变有效的原癌基因,以干细胞因子(SCF)为配体,通过破坏细胞生长调控导致肿瘤的发生[23]。KIT 在人类肿瘤中的确切作用仍不明了,但目前从文献来看,差异主要取决于肿瘤类型。如KIT 在小细胞肺癌、白血病、结肠癌和成神经细胞瘤中高表达或突变[24-27];而在乳腺癌和黑色素瘤中表达缺失[28-29]。关于KIT 在甲状腺癌中的研究较少。
Mazzanti 等[30]在2004年通过基因芯片技术在数千个基因中筛选出KIT 基因,发现与甲状腺良性结节相比,KIT 在甲状腺癌组织中是表达下调最明显的蛋白编码基因之一。近来,有研究进一步证实了KIT 在甲状腺正常组织和甲状腺癌组织中的表达,发现与甲状腺正常组织相比,KIT 在甲状腺癌中表达下调[31-32],与本研究结果相一致。Tomei 和Franceschi 等[11,33]根据研究结果推测KIT 与其配体的结合可以调控甲状腺滤泡上皮的分化和生长,KIT表达的缺失可能会导致甲状腺结节向恶性转化,并认为KIT 可作为甲状腺细针穿刺细胞学诊断的分子标志物。更有一些研究报道了引起KIT 在甲状腺癌中表达下调的作用机制,包括特异性microRNA(miR-146b、miR-221 和miR-222) 的表达失调、启动子高甲基化以及lncRNA 的竞争性内源性学说[7,29,34]。目前,甲状腺细针穿刺细胞学检查仍然是术前评估甲状腺结节良恶性的最可靠、经济、安全的检查手段,有助于减少不必要的外科手术[35-37]。众所周知,BRAF V600E 是甲状腺乳头状癌中广泛应用的分子标志物之一,检测BRAF V600E 有无突变可使术前检查准确率提高20%~30%[38]。但依然有部分患者的诊断具有不确定性,原因可能是该部分患者中不存在BRAF V600E 突变,提示识别甲状腺癌中其他的分子标志物具有重要意义[11]。Tomei 等[11]研究发现在甲状腺细针穿刺细胞学样本中联合检测BRAF V600E 突变和KIT表达,可使诊断准确率提高15%。Panebianco 等[35]研究报道了在甲状腺细针穿刺细胞学样本中联合检测4 种分子标志物(KIT、TC1、miR-222、miR-146b)比单纯检测BRAF V600E 突变更有助于恶性结节的诊断,推荐用于临床上无法诊断甲状腺结节良/恶性的病例中。由此可见,KIT 不仅可作为甲状腺癌的预后风险基因,检测其蛋白表达还有助于甲状腺结节的术前诊断。
综合本研究结果及文献资料,KIT 基因可被用于甲状腺癌诊断的分子标志物、治疗靶点及预后风险评估。
猜你喜欢
昆明医科大学学报(2021年10期)2021-12-02 03:24:14
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:12
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:46
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:24
汉字汉语研究(2020年2期)2020-08-13 07:52:48
电子制作(2019年22期)2020-01-14 03:16:24
疯狂英语·新读写(2018年3期)2018-11-29 22:37:11
中国卫生标准管理(2015年2期)2016-01-15 00:31:34
中国中西医结合外科杂志(2013年3期)2013-03-11 20:05:01
中国医学科学院学报(2011年1期)2011-03-25 13:58:42
