
原发性肝癌中PD-L1表达的调控机制
2021-12-07 00:52:14李圣豪石新丽
肿瘤防治研究 2021年11期
李圣豪,石新丽
0 引言
原发性肝癌是全球第三大癌症死亡原因,其中肝细胞癌(hepatocellular carcinoma,HCC)约占原发性肝癌的75%~85%[1]。几十年来,有效改善晚期肝癌预后的疗法较少。酪氨酸蛋白激酶抑制剂如索拉非尼、瑞戈非尼和乐伐替尼被认为是最有效的靶向药物,并且是晚期肝癌患者的唯一有效疗法[2]。然而,最近一项关于索拉非尼在亚太地区晚期肝癌患者中的有效性和安全性临床试验证明,索拉非尼只能将晚期HCC患者的生存期延长3月,并且出现了耐药的问题[3]。
免疫治疗被誉为肿瘤学的重大突破。最近研究发现靶向程序性细胞死亡受体配体-1(programmed cell death ligand 1,PD-L1)/程序性细胞死亡受体-1(programmed cell death 1,PD-1)的药物在HCC治疗中具有显著的抗肿瘤作用[4],在HCC肿瘤微环境中,PD-L1主要在肿瘤细胞和宿主免疫细胞中表达。PD-L1表达增加导致抗肿瘤T细胞迁移、增殖和细胞毒性分泌的抑制[5]。因此,明确PD-L1在肝癌中的调控机制对于免疫治疗至关重要。
1 PD-L1在宿主免疫细胞上的表达
1.1 骨髓来源抑制细胞(myeloid-derived suppressor cells,MDSCs)
骨髓来源的抑制细胞在免疫调节中起重要作用,但MDSCs在肝细胞癌患者中的免疫抑制功能尚未阐明。高表达集落刺激因子1(colony stimulating factor 1,M-CSF)和血管内皮生长因子A(vascular endothelial growth factor A,VEGFA)的肝癌细胞系显著诱导MDSCs PD-L1的表达[6]。进一步研究发现MDSCs有助于肿瘤免疫抑制微环境形成。HCC患者的肿瘤浸润性CD11b+CD33+HLADR-MDSCs可以有效抑制CD8+T细胞的增殖。进一步研究发现肝细胞周期蛋白依赖性激酶(cyclin dependent kinase,CCRK)通过激活EZH2/核转录因子-κB(nuclear transcription factor-κB,NF-κB)/IL-6级联反应导致MDSCs积累[7]。相反,值得注意的是,肿瘤性CCRK耗竭上调了PD-L1表达并增加了肿瘤内CD8+T细胞,从而增强了PD-L1抗体治疗肝癌的作用。
1.2 肿瘤相关巨噬细胞
巨噬细胞分化与肝细胞癌的发生密切相关。巨噬细胞分化相关基因(macrophage differentiation-associated genes,MDGs)中的CDC42与M2巨噬细胞标志物和免疫检查点呈正相关,CDC42的表达与Wnt信号通路密切相关。研究表明CDC42是肝癌中重要的MDGs,并且被证明是研究肝癌中巨噬细胞分化的新基因[8]。类赖氨酰氧化酶4(lysyl oxidase like 4,LOXL4)是一种胺氧化酶,主要参与细胞外基质重塑,并在肝细胞癌组织中高表达,但其在介导肝癌发生中的功能未知。LOXL4对巨噬细胞的免疫抑制功能主要依赖于IFN介导的STATs依赖性PD-L1活化,从而进一步抑制CD8+T细胞的功能[9]。
研究发现肝癌外周血单核细胞中的肿瘤浸润性T细胞的O型蛋白酪氨酸磷酸酶受体(protein tyrosine phosphatase receptor type o,PTPRO)显著降低,并且PTPRO与肝癌外周血单核细胞和肿瘤相关巨噬细胞中PD-L1表达的增加有关。血清IL-6通过激活STAT3/c-MYC/miR-25-3p轴,降低PTPRO的表达,引起PD-L1诱导的免疫抑制来促进肿瘤生长[10]。HCC细胞发生内质网应激,并以释放外泌体的形式促进免疫抑制的形成,外泌体通过miR-23a-PTEN-AKT途径上调巨噬细胞中PD-L1的表达,巨噬细胞进一步抑制T细胞功能[11]。这些研究为深入了解肿瘤细胞的肿瘤免疫逃避机制提供了依据。
1.3 单核细胞
活化单核细胞释放的自分泌肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)和IL-10刺激单核细胞自身PD-L1的表达。PD-L1阳性单核细胞可有效抑制肿瘤特异性T细胞免疫,并促进人体内肿瘤的生长。肿瘤来源的可溶性因子透明质酸片段诱导了肿瘤相关的单核细胞中关键糖酵解酶PFKFB3的上调,该酶通过激活细胞中的NF-κB信号促进PD-L1的表达[12]。该机制揭示了细胞代谢可以调节肿瘤微环境中单核细胞的促肿瘤功能。
在肿瘤微环境中,免疫逃逸的关键驱动因素肿瘤细胞内源性骨桥蛋白(osteopontin,OPN)具有致癌活性,肝癌组织中OPN和PD-L1表达以及肿瘤相关巨噬细胞(tumor-associated macrophage,TAM)浸润之间存在正相关。OPN促进了巨噬细胞趋化性迁移和替代激活,并通过激活巨噬细胞中的CSF1-CSF1R途径促进肿瘤细胞中PD-L1的表达[13],阻断该途径可防止TAM转运,从而增强免疫检查点抑制剂治疗HCC的疗效。
M1型巨噬细胞被认为具有杀伤肿瘤作用,但一些研究报道了其对肿瘤的促进作用。CD68+HLADR+M1型巨噬细胞的浸润与HCC细胞中PD-L1表达水平有关,M1型巨噬细胞分泌IL-1β可诱导肿瘤细胞NF-κB p65和转录调节因子-1(interferon regulatory factor-1,IRF-1)的表达,促进PD-L1表达[14]。
PD-L1水平的升高代表了肿瘤细胞响应内源性抗肿瘤活性而产生的适应性免疫抵抗机制,肿瘤细胞PD-L1的上调主要是由肿瘤微环境中存在的活化CD8+T细胞诱导的,它是HCC有利的预后因素[15]。也有研究认为PD-L1能下调肿瘤微环境中T细胞活化相关的基因。高表达PD-L1的小鼠肝癌细胞系BNL-MEA与CD8+T细胞共培养可降低T细胞增殖以及细胞因子IFN-γ和TNF-α的表达[16]。虽然表达PD-L1的肿瘤显示出对抗PD-1治疗更好的反应,但是CD8+T细胞的耗竭消除了抗PD-1的抗肿瘤效果。
2 PD-L1的表观遗传调控
果蝇zeste基因增强子的人类同源物2(enhancer of zeste homolog 2,EZH2)以IFN-γ依赖性方式负调控肝癌细胞系PD-L1的表达。EZH2可以通过上调CD274启动子上的组蛋白H3的N端第27位赖氨酸三甲基化水平(编码PD-L1)和干扰素调节因子1(IRF1)来抑制PD-L1表达[17],EZH2是免疫联合治疗HCC的潜在治疗靶点。
3 PD-L1转录调控
3.1 STAT3信号通路
研究发现信号转导与转录激活因子3(signal transducer and activator of transcription 3,STAT3)与PD-L1启动子结合,转录调控PD-L1表达[18]。
STAT3活性降低,则降低IFN-γ诱导的PD-L1表达,并恢复T细胞杀伤肿瘤细胞功能[19]。另外,STAT3上游分子通路对STAT3的磷酸化和糖基化修饰,也影响PD-L1的表达。D蛋白酪氨酸磷酸酶受体(protein tyrosine phosphatase receptor type D,PTPRD)是一种重要的肿瘤抑制基因,可通过抑制STAT3磷酸化,抑制PD-L1的表达[20]。高尔基膜蛋白1(Golgi membrane protein 1,GOLM1)作为一种致癌基因,通过选择性地与EGFR结合促进肝癌的生长和转移。另外,研究还发现GOLM1通过增强EGFR的水平来促进STAT3的磷酸化,进而上调PD-L1的转录表达[21]。Toll样受体 9(Tolllike receptors 9,TLR9)负调节PARP1表达,从而介导STAT3多聚ADP核糖基化修饰(Poly(ADPribosyl)ation,PARylation)的减少和STAT3 Tyr705磷酸化的增加,促进PD-L1转录[22]。
3.2 MAPK信号通路
有丝分裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路与肝癌中PD-L1基因的表达有关。表皮生长因子(epidermal growth factor,EGF)或IFN-γ的刺激可增加PD-L1转录和蛋白水平的表达。表皮生长因子受体、丝裂原活化蛋白激酶激酶1和丝裂原活化蛋白激酶激酶2阻断,可抑制EGF和IFN-γ诱导的PD-L1转录和蛋白表达水平上调。IFN-γ增加了PD-L1的转录活性,而MAPK信号转导增加了PD-L1 mRNA的稳定性[23]。另外MAPK还受到其他信号通路的调控,受体酪氨酸激酶C-MET与肝细胞生长因子(hepatocyte growth factor,HGF)结合形成二聚体,导致C-MET羧基末端酪氨酸磷酸化,进而导致MAPK和PI3K活化。HGF诱导的C-MET活化发生在PD-1/PD-L1信号通路的活化过程中,因此在HCC中C-MET调节PD-L1的表达[24]。
3.3 IFN-γ信号通路
肿瘤浸润性T细胞释放IFN-γ诱导PD-L1表达[25]。IFN-γ通过上调小鼠和人HCC细胞中IRF-1的表达诱导PD-L1转录和蛋白水平表达[26]。研究也发现人肝癌细胞系中IFN-γ主要依赖JAK/STAT1/IRF1途径诱导PD-L1表达[27]。转录因子IRF-1和IRF-2信号通路均调节HCC细胞中的PD-L1的表达。IRF-2过表达可以抑制IFN-γ诱导的PD-L1启动子活性和蛋白水平。而IRF-1拮抗IRF-2与PD-L1中的IRE启动子结合,为HCC免疫检查点抑制剂治疗中PD-1/PD-L1途径的调控提供了新的启示。研究发现TNF-α通过上调IFN-γ受体的表达来增强IFN-γ信号转导。另外,研究发现IFN刺激基因12a(IFN stimulates gene 12a,ISG12a)通过抑制Axin泛素化降解来促进β-catenin蛋白酶体降解,从而抑制了经典的Wnt/β-catenin信号转导途径[28]。β-catenin被鉴定为PD-L1的转录因子。ISG12a抑制Wnt/β-catenin信号转导可抑制免疫检查点PD-L1的表达,从而使NK细胞对癌细胞杀伤敏感,这项研究揭示了IFN抗癌作用的潜在机制。
3.4 雄激素受体(androgen receptor,AR)信号通路
雄激素受体作为PD-L1的转录抑制因子负调控PD-L1[29]。体外实验发现肝癌细胞中AR的过表达增强了CD8+T的功能,AR低表达的肿瘤对PD-L1抑制剂的反应较好。因此,AR抑制PD-L1的表达可能与HCC中的性别差异有关。另外有研究发现,羟类固醇17-β脱氢酶6(hydroxysteroid 17-β dehydrogenase 6,HSD17B6)是参与合成二氢睾丸激素的关键蛋白,在HCC的发生和发展中发挥重要的作用。HSD17B6可通过产生双氢睾酮(dihydrotestosterone,DHT)抑制TGF-β1和PD-L1的表达[30]。
3.5 其他特异性转录调控
氧化应激反应1(oxidative stress response 1,OXSR1)与部分肿瘤的恶性进展密切相关。OXSR1在肝癌组织中上调,促进肿瘤细胞的增殖、迁移和侵袭。此外,OXSR1的表达水平与HCC中肿瘤浸润免疫细胞的浸润水平和PD-L1表达呈正相关[31]。MYC原癌基因上调淋巴瘤中PD-L1的表达,HCC细胞中敲低MYC,增加了PD-L1 mRNA和蛋白质水平的表达[32]。MYC抑制信号转导及转录激活子-1 (signal transducer and activator of transcription 1,STAT1) 的表达,STAT1是IFN-γ信号通路的重要组成部分,IFN-γ刺激HCC细胞促进PD-L1表达。Hippo信号通路在多种癌症类型中失活,导致定位在细胞质当中的Yes1相关转录调节因子(Yes-associated transcriptional regulator,YAP1)进入细胞核,调控下游相关基因的转录。研究发现,YAP1抑制剂可以降低肝癌组织PD-L1的表达,破坏肿瘤的免疫抑制微环境,从而改善HCC的治疗效果[33]。
4 转录后调控PD-L1的表达
4.1 MicroRNAs对PD-L1的调节
MicroRNA-1(miR-1)是一种肿瘤抑制性microRNA,直接调控PD-L1表达。miR-1的缺失促进对索拉非尼耐药的肝癌细胞中PD-L1的表达[34]。癌基因转录因子核因子E2相关因子2(Nuclear factor,erythroid 2 like 2,NRF-2)可抑制miR-1的表达,NRF-2/miR-1/PD-L1调控轴有助于索拉非尼耐药肝癌细胞耐药性的维持和发展,并为克服HCC中索拉非尼耐药性提供了潜在的治疗靶点。另外研究发现miR-155-5p和miR-194-5p可通过X失活的特异性转录本(X inactive specific transcript,XIST)上调PD-L1的表达[35]。
4.2 LncRNAs对PD-L1的调节
在HCC中,长链非编码RNA PCED1B-AS1(PCED1B antisense RNA 1,PCED1B-AS1)和hsamiR-194-5p表达上调。PCED1B-AS1与PD-Ls正相关,而与hsa-miR-194-5p负相关。PCED1B-AS1与抑制PD-Ls表达的hsa-mir-194-5p相互作用,增强PD-Ls的表达[36]。癌症易感性候选物11(cancer susceptibility 11,CASC11)募集真核翻译起始因子4A3(eukaryotic translation initiation factor 4A3,EIF4A3),并增强E2F1 mRNA的稳定性,促进NF-κB信号和PI3K/AKT/mTOR通路的激活,调节PD-L1的表达[37]。研究还表明,肝癌中长链非编码RNA MIAT的表达与PD-1、PD-L1和CTLA4等免疫检查点分子的表达呈正相关[38]。
LINC00657在HCC组织和细胞系中过表达,与预后不良有关。LINC00657通过降低miR-42调节PD-L1的表达,PD-L1的3'UTR与miR-424高度保守,miR-424可显著抑制PD-L1的mRNA和蛋白表达水平[39]。长链非编码RNA KCNQ1重叠转录本1(KCNQ1 overlapping transcript 1,KCNQ1OT1)已被证明与部分肿瘤细胞的耐药性有关。KCNQ1OT1充当miR 506的竞争性内源RNA,并在索拉非尼耐药的肝癌细胞中促进PD-L1的表达[40]。
5 翻译后修饰对PD-L1的调控作用
EGF刺激促进了HCC细胞中PD-L1 mRNA和蛋白的表达。EGF处理导致PD-L1启动子处的H3-Thr11磷酸化增强,抑制表皮生长因子受体可以逆转EGF诱导的PD-L1 mRNA和蛋白的表达。抑制丙酮酸激酶同工型M2(pyruvate kinase M2,PKM2)则显著阻止EGF诱导的PD-L1表达和H3-Thr11的磷酸化[41]。肝癌患者的肿瘤组织样本中观察到聚腺苷酸二磷酸核糖转移酶-1(poly(ADPribose)polymerase-1,PARP-1)和磷酸化的糖原合成酶激酶3β(p-glycogen synthase kinase 3 beta,p-GSK3β)或PD-L1表达之间呈负相关,抑制PARP1活性可以增强GSK3β磷酸化,上调PD-L1表达,并最终抑制T细胞浸润[42]。
6 免疫检查点抑制剂的临床应用
肿瘤免疫治疗的特点在于激发特异性免疫反应。国产卡瑞利珠单抗单药用于晚期肝癌的治疗,客观缓解率14.7%,6月生存率74.4%,12月生存率55.9%,中位生存期13.8月[43]。替雷利珠单抗单药治疗晚期二线肝细胞癌患者的ORR高达18.8%,DCR达56.3%,在同类药物中处于领先水平[44]。另外,PD-L1单抗在肝胆肿瘤治疗中也开展了很多临床试验。一项关于阿替利珠单抗联合贝伐单抗一线治疗晚期肝细胞肝癌患者的研究结果显示,索拉非尼组mOS为13.2月,联合组可使死亡风险降低42%;联合组的mPFS为6.8月,索拉非尼组为4.3月,疾病进展风险降低41%。此外,在中国患者中带来更大获益,降低了56%的死亡风险[45]。该联合方案不仅被NCCN指南纳入肝癌患者的一线治疗,也被FDA批准为肝癌史上首个免疫联合的一线治疗方案。2020 CSCO指南将阿替利珠单抗联合贝伐单抗组合纳入晚期肝癌一线治疗的Ⅰ级推荐。
7 总结与展望
综上所述,肝癌免疫微环境中PD-L1在多种细胞上表达,包括巨噬细胞、单核细胞和肿瘤细胞等。PD-L1的表达可明显抑制效应T细胞的抗肿瘤作用。目前针对PD-L1的研究主要集中在免疫治疗方面,但也有研究发现,PD-L1也可不依赖PD-1/PD-L1途径,发挥促进肿瘤生长的作用。另外PD-L1表达的调控受到多种因素的调控,包括转录、转录后翻译和翻译后修饰等多层面的调控,而且不同层次和信号通路之间交叉调控,机制复杂,见表1。PD-L1阳性的患者可在免疫检查点抑制剂的治疗中获益,但是PD-L1阳性的患者并未在治疗中完全有效。相反,PD-L1阴性的患者也可以在免疫检查点抑制剂治疗中获益。免疫检查点抑制剂治疗失败的原因可能是肿瘤细胞免疫原性的突变、肿瘤细胞上多种免疫检查点的配体的表达以及T细胞的浸润缺陷。因此,免疫检查点抑制剂的联合治疗策略,为免疫检查点抑制剂的应用拓展了新空间,同时也显现出非常显著的治疗效果,最终将为患者带来更多的选择,延长患者生存期。
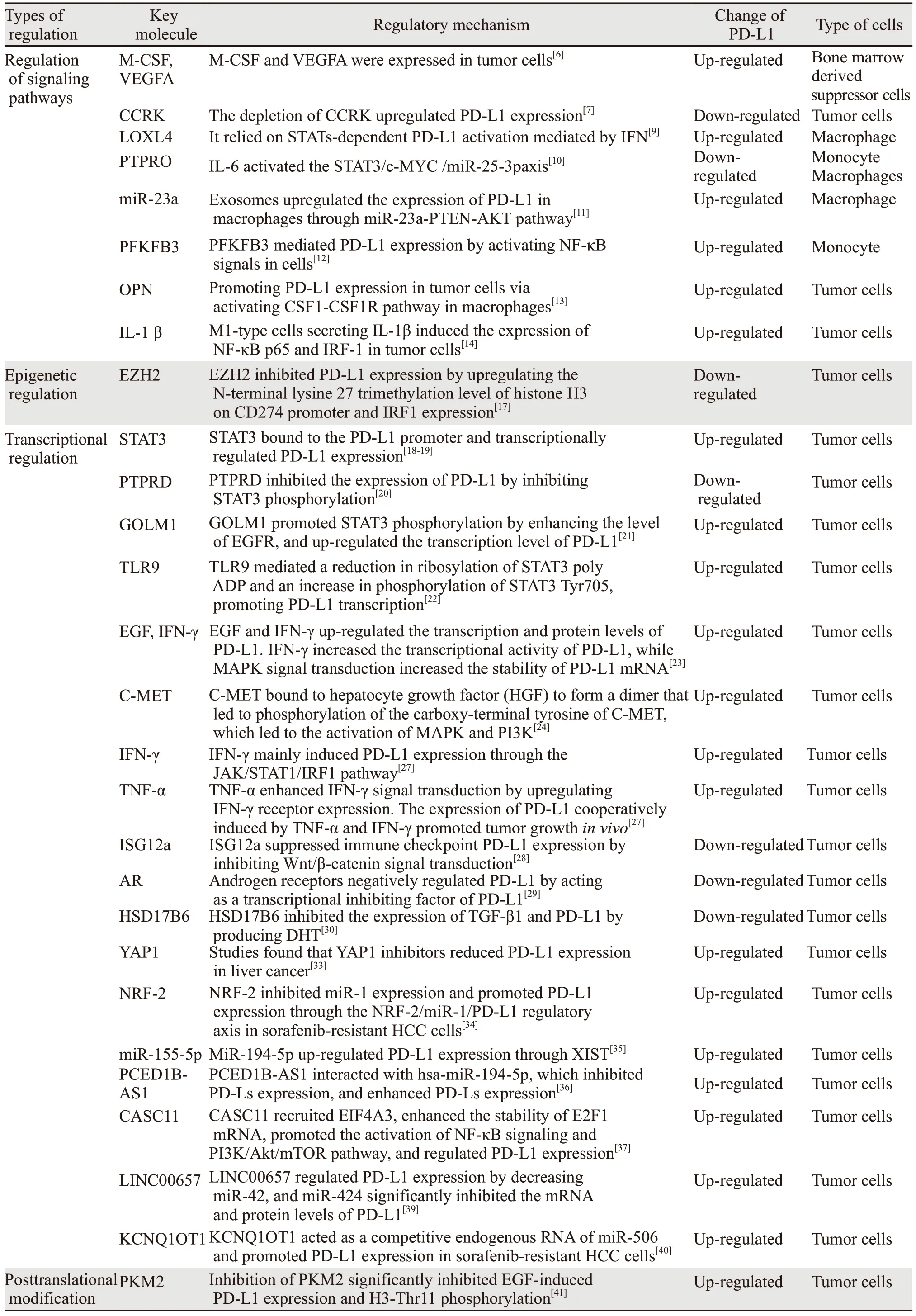
表1 肝癌PD-L1的调控机制Table 1 Regulatory mechanism of PD-L1 in liver cancer
猜你喜欢
计算机系统应用(2022年4期)2022-05-10 08:41:10
天津医科大学学报(2021年4期)2021-08-21 02:14:52
天津医科大学学报(2019年3期)2019-08-13 06:53:08
国际呼吸杂志(2019年4期)2019-03-12 01:08:18
上海农业学报(2017年3期)2017-04-10 12:39:26
现代计算机(2015年31期)2015-09-28 05:31:51
肿瘤预防与治疗(2015年1期)2015-09-26 07:26:20
中国当代医药(2015年16期)2015-03-01 02:03:13
中国当代医药(2015年16期)2015-03-01 02:03:11
癌变·畸变·突变(2015年4期)2015-02-27 06:15:25
