
Thermal Unfolding and Aggregation Process of Recombinant Staphylococcal Enterotoxin M (rSEM) Associated with Potential Toxin Responsible for Staphylococcal Food Poisoning
2021-12-02 09:20LIUJiTIANWanfanTANGJunniCHENJuanZHAOYanyingYUJicheng
食品科学 2021年21期
LIU Ji, TIAN Wanfan, TANG Junni,*, CHEN Juan, ZHAO Yanying, YU Jicheng
(1. College of Food Sciences and Technology, Southwest Minzu University, Chengdu 610041, China;2. College of Animal and Veterinary Sciences, Southwest Minzu University, Chengdu 610041, China;3. Key Laboratory of Biotechnology and Bioresources Utilization, Dalian Minzu University, Dalian 116600, China)
Abstract: Thermal processing is one of the most useful tools to reduce the amounts of bacteria and toxins that may potentially be present in processed foods. Staphylococcal enterotoxin M is a newly identified group V superantigen with mild emetic activity and has the potential risk of causing staphylococcal food poisoning. In this study, the heat-induced conformational changes of recombinant staphylococcal enterotoxin M (rSEM) were identified by circular dichroism (CD),fluorescence spectroscopy and sodium dodecyl sulfate/native-polyacrylamide gel electrophoresis (SDS/native-PAGE).Below 40 ℃, rSEM had a well-folded structure with high contents of α-helix (17%), β-sheet (32%) and β-turn (21%) and the single tryptophan residue at position 121 (Trp121) on its molecular surface was found be located in the hydrophobic environment of the β-grasp domain. As the heating temperature increased from 42 to 55 ℃, α-helix content decreased,and β-sheet/turn contents increased to compensate for this. The aggregation state of the protein did not change markedly,while a distinct blue shift in the fluorescence emission maxima was observed accompanied by the reverse S-shaped curve for the ratio of fluorescence intensities at 350 and 340 nm, indicating the formation of an alternatively folded state, namely the intermediate state (IS). When the temperature was above 55 ℃, the secondary structure elements persisted even upon heating to 90 ℃. Meanwhile, upon heating from 65 to 80 ℃, the ratio of fluorescence intensities at 350 and 340 nm didn’t show that the protein was in the completely unfolded state. These results on well-folded secondary structure and tertiary structure variations imply stable integrated architecture of the protein and that the flexible β-grasp domain is responsible for binding to the diverse major histocompatibility complex (MHC) alleles. Besides, with increasing temperature from 70 ℃, the aggregation level increased visibly and reached its maximum at 90 ℃. Taken together, all data showed that the β-sheet/turn structure of rSEM and the formation of IS and the aggregation state were predominantly responsible for the structural stability at high temperature. Understanding the heat-induced unfolding process of rSEM will help in clarifying its heat resistance mechanism. In the future, using this method to study the heat inactivation mechanism of other types of staphylococcal enterotoxin will help in improving the food production process.
Keywords: staphylococcal enterotoxin M; circular dichroism; fluorescence; sodium dodecyl sulfate/native polyacrylamide gel electrophoresis ; heat inactivation
Staphylococcal entertoxins (SEs) are known as virulence factor due to their causation of staphylococcal food poisoning (SFP)[1]. As highly successful foodborne toxins with highly heat/proteolysis resistance, acid tolerance and desiccation-tolerance[2-5], they may put consumers at the risk of SFP. Moreover, their biological toxicity and environmental stability have resulted in some entertoxins being categorized as select agents of bioterroris[6]. SEs are also known as superantigens due to their ability to nonspecifically stimulate a large population of T cell[7]by means of binding to major histocompatibility complex (MHC) II molecules on antigen presenting cells and binding to the T cell receptor on T cells. The staphylococcal superantigens[8]include TSST-1, SEs (serotypes A, Bn, Cn(where n refers to multiple variants), D, E, and G) and SE-like (SE-l)superantigens (serotypes H, I and J to X). All these superantigens share a common three dimensional fold: an amino-terminal oligosaccharide/oligonucleotide binding (O/B)fold comprised of aβ-barrel domain, a carboxy-terminalβ-grasp domain made of antiparallelβ-strands and a central,diagonalα-helix bridged crossing above-mentioned two domains. Based on small variations in this common core structure, superantigens can be divided into five evolutionary groups (I-V)[9], and staphylococcal enterotoxins M (SEM)belongs to the evolutionary group V. In fact, group V superantigens are the last described group and contain many of the recently discovered superantigens[8]. These include SE-lK to SE-lM, SE-lQ and SE-lV, and lack the emetic activity cystine loop but also contain both the low- and high-affinity MHC II binding sites exactly as group III and IV. Recently,mild emetic potential of SEM has been demonstrated[10],which indicated that SEM might be associated with SFP.SEM, like other group V superantigens, also have an additional 15-amino-acid insert. The insert, named theα3-β8 loop, exists between the thirdα-helix and theβ-strand 8. This loop appears to be critical for the specificity of the interaction of the superantigens with their respective Vβ-T cell receptors (TCRs)[11]. The research about SEM dated back to 2001, Jarraud et al.[12]were the first to recognize that enterotoxins gene cluster (egc) is the location ofsemgene. Furthermore, they purified rSEM fromE. colifor T cell proliferation assays. Subsequent work by Pan Yingqiu et al.[13]also identified bioactivity of rSEM in manner of prokaryotic expression of recombinant protein. Later work by Omoe et al.[10]established emetic potentials of rSEM fromE. coli. In addition, numerous studies have shown that reliable results could be obtained by prokaryotic expression recombinant staphylococcal entertoxins (rSEs) inE. coli.Taken together, all the data demonstrate the structural and functional similarity between rSEs inE. coliand natural SEs inS. aureus. Therefore, prokaryotic expression of rSEs inE. coliis an universally accepted method to investigate structural and functional relationship of SEs inS. aureus.
Thermal inactivation is a common method to neutralize pathogenic bacteria and their toxins during food processing.The bacteria will be inactivated during this process in most cases, but the preformed toxins such as SEs, may persist and cause SFP or other food-borne diseases. Thus, in the effort to establish safer guidelines for food preparation, thermostability and structural changes of enterotoxins during heat treatment should be investigated, which is requisite to improve food quality and safety. Recently, a research on the thermal stability of group III superantigens involving in staphylococcal entertoxin A (SEA), staphylococcal entertoxin E (SEE) and staphylococcal entertoxin H (SEH) suggested that the family of SEs have different ability to withstand heat[14].Therefore, the exact profile of heat inactivation for those specific SEs causing SFP needs to be considered individually to improve food safety. Although some publications have addressed the structure[15-17], toxicity[18-20], superantigen activity[21-23]and highlysensitive detection of entertoxins[24-26], to date the mechanism of thermal stability remains far too unclear. Biological functions of proteins depend on the correct folding of their native structure and loss of compact folded structure leads to an unfolded, inactive state. Therefore, the study of factors affecting conformational stability is important both from the academic and applied points of view.
The exact profile of unfolding and aggregation process for SEM induced by heat has not been characterized.Therefore, in this study we applied a combination of circular dichroism (CD), fluorescence spectroscopy and sodium dodecyl sulfate (SDS)/native-polyacrylamide gel electrophoresis (PAGE) to describe the thermal-induced structural changes of rSEM at the secondary, tertiary and quaternary structural level.
1 Materials and Methods
1.1 Materials and reagents
Staphylococcus aureus(SA003) was kept in our laboratory and used for the total DNA extraction.Escherichia coli(E. coli) DH5α andE. coliRosetta (DE3) were purchased from Tiangen Biotech Co., Ltd. (Beijing, China). The plasmid pET-28-a(+) was purchased from Novagen Pharmaceutical Company INC. (Germany). The restriction endonucleases NdeI, XhoI and Pfu DNA Polymerase, T4 DNA ligase was purchased from TaKaRa (Dalian). DNA sequencing was executed by the Tsingke Biological Technology Company(ChengDu). Ni2+-NTA-Sepharose was purchased from GE Healthcare (USA); Thrombin was purchased from Sigma-Aldrich (USA); and the culture media Luria-Bertani (LB) was self-prepared.
1.2 Instruments and equipments
Amicon®Ultra-4 centrifugal filter devices, UV-2550 UVVis spectrophotometer Japan Shimadzu Company; AVIV Model 400 CD spectrophotometer US Aviv Biomedical Inc.; F-7500 fluorescence spectrophotometer Japan Hitachi Ltd..
1.3 Methods
1.3.1 DNA extraction, PCR amplification, expression and purification of rSEM protein
Genomic DNA was purified and applied as template for amplification ofsemgene by polymerase chain reaction (PCR)with forward and reverse primers below.
Forward primer: 5′-ATGAAAAGAATACTTATCATTG-3′
Reverse primer: 5′-TCAACTTTCGTCCTTATAAG-3′
These primers were based on the sequences ofsemgene truncated signal peptide fromS. aureus04-02981 (GenBank accession No. CP001844.1).
In brief, the recombinant strains were incubated at 37 ℃for 12 h LB medium with 30 mg/mL kanamycin. The cultures were diluted (1:100) in the same mediumand grown at 37℃ to reach an absorbance of approximately 0.6 at 600 nm wavelength. The microbial content was examined by the changes of absorbance at 600 nm wavelength in culture liquid spectrophotometrically. The expression of the recombinant proteins was induced by 1 mmol/L isopropyl-β-D-thiogalactoside at 37 ℃ and the cells, after 6 h of induction,were harvested by centrifugation at 4 000 r/min for 10 min at 4 ℃.The pellet was washed and resuspended in buffer A (20 mmol/L Tris-HCl, 0.5 mol/L NaCl, 5 mmol/L imidazole, pH 8.0),and then the cells were lysed by an ultrasonic oscillator.After centrifugation at 10 000 r/min for 20 min, the extracted recombinant soluble proteins were purified by Ni-chelating column. The column was washed by buffer A (20 mmol/L Tris-HCl, 500 mmol/L NaCl, pH 8.0), buffer B (20 mmol/L Tris-HCl, 500 mmol/L NaCl, 100 mmol/L imidazole, pH 8.0)and buffer C (20 mmol/L Tris-HCl, 500 mmol/L NaCl,500 mmol/L imidazole, pH 8.0) consecutively. The major fraction of His-tagged fusion protein that specifically bind to Ni2+was eluted by buffer C. The purity of the eluted protein was evaluated by SDS-PAGE and enzyme-linked immunosorbent assay established in our laboratory[26].
1.3.2 Thrombin digestion and purification of rSEM
The fusion protein purified from Ni2+-chelating column with imidazole would undergo solvent exchange by centrifugal filter devices (Amicon®Ultra-4) before thrombin digestion. Thereafter, the fusion proteins His-rSEM was digested by thrombin and purified with DEAE-Sephadex ionexchange column. The wanted protein rSEM was harvested in 1 mmol/L sodium phosphate buffer (pH 7.0) supplemented with 1 mmol/L ethylene diamine tetraacetic acid (EDTA).Finally, the wanted protein rSEM was ultra-filtrated by centrifugal filter devices (Amicon®Ultra-4) in 1 mmol/L sodium phosphate buffer pH 7.0 for the subsequent gel electrophoresis assay and spectroscopic measurement.
1.3.3 SDS-PAGE and native-PAGE
Electrophoresis in SDS-PAGE was conducted according to the Laemmli’s method[27], using a 4% (m/V) stacking gel and a 12.5% (m/V) resolving gel at a constant current of 10 mA for 2 h. The molecular weight of purified protein was estimated by comparison with migration ratio of standard molecular weight. Electrophoresis in native-PAGE was conducted according to the Davis’s method[28], using a 4%(m/V) stacking gel and a 12.5% (m/V) resolving gel at a constant current of 10 mA for 8 h at 4 ℃. For native-PAGE,the molecular weight of purified protein was estimated by comparison with migration ratio of standard Bovine Serum Albumin (fraction V). The gel was stained with 0.1%(m/V) Coomassie Brilliant Blue R-250 in 40% (V/V) methanol and 7% (V/V) acetic acid for 1 h at room temperature and destained with 40% (V/V) methanol with 7% (V/V) acetic acid for 12 h. Before SDS-PAGE or native-PAGE was carried out, the proteins were diluted into reducing or non-reducing electrophoretic loading buffer, respectively. For reducing loading buffer 5×, there were 2.9 mmol/Lβ-mercaptoethanol and 13.9 mmol/L SDS in solution; For non-reducing loading buffer 5 ×, there was only 13.9 mmol/L SDS in solution.
1.3.4 Thermal stability analysis of rSEM by SDS-PAGE
The purified rSEM was centrifuged to get rid of any aggregated protein before sampling, and then diluted with 10 volume sodium phosphate buffer (1 mmol/L, pH 7.0) to a protein concentration of 8 μmol/L. rSEM (1 000 μL) was added in Eppendorf tube and then subjected to consecutive heating from 30 to 90 ℃ with an increment of 10 ℃ for 10 min, respectively. Following thermal challenge of proteins at the specified temperature intervals, an aliquot of each protein was allowed to thaw on ice, transferred into a new Eppendorf tube and dissolved into loading buffer 5 × containing 2.9 mmol/Lβ-mercaptoethanol and 13.9 mmol/L SDS by vortex. Finally, the protein samples (6 μg) were loaded to 12.5% SDS-PAGE.
1.3.5 CD measurements
CD spectroscopy measurements were performed on an AVIV Model 400 CD spectrophotometer equipped with a Peltier thermo-stated cell holder. A 2 mm quartz cuvette was placed in above-mentioned cell holder and regulated by Peltier thermostat. The rSEM was diluted as described in section 1.3.4, then heated in the quartz cuvette in-site and measured at 190-260 nm. The thermal unfolding of CD spectra were monitored along with the heating procedure from 25 to 90 ℃ with appropriate temperature intervals and equilibrated at each temperature for 10 min before the CD spectra were recorded. Every spectrum was the means of three scans and the spectrum of blank buffer was subtracted. Results were obtained in millidegrees/theta(machine units) and subsequently converted to the mean residue ellipticity (MRE) based on 217 amino acid residues and a molecular weight of 24.841 kDa. The concentration of total protein was 0.084 mg/mL, determined by UV/Vis absorbance spectroscopy using the extinction coefficient at 29 340 L/(mol·cm). The CD spectra were expressed in terms of molar ellipticity per residue, defined by formula (1) and (2).

Whereθis the CD millidegrees/theta (machine units);ρis the protein concentration/(mg/mL);lis the length of the light beam’s path through the sample/cm;Mmrwis the mean residue weight (MRW)/Da.
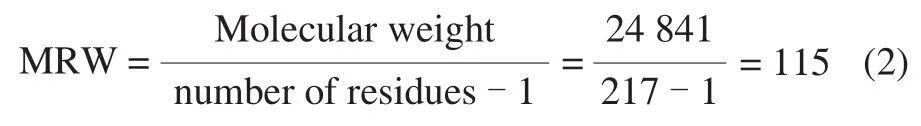
To gain information on the secondary structure, the CD spectra of the rSEM was analyzed using the program packages DICHROWEB[29]. DICHROWEB is a Web server set up at the Department of Crystallography of the Institute of Structure and Molecular Biology, Birkbeck College,University of London, UK. It is publicly available for nonprofit organizations at http://dichroweb.cryst.bbk.ac.uk. from a reference set[30]of proteins with known structure and known CD spectra, using the singular value deconvolution algorithm and variable selection procedures, the programs calculate the fraction of each secondary structure motif that contributes to the spectrum of protein.
1.3.6 Fluorescence measurements
The sample of rSEM was diluted and heated as described in section 1.3.5. The rSEM (0.56 μmol/L) was used for all the following fluorescence measurement on the F-7500 fluorescence spectrophotometer. The intrinsic fluorescence measurements of rSEM excited at 278 nm were recorded.Excitation slit and emission slit were set as 5 nm. rSEM(800 μL) was heat-treated in-site in 1 cm pathlength cuvette in the fluorescence spectrophotometer equipped with a temperature-controlled water-bath. The emission spectra were recorded between 300-400 nm and every spectrum was the average of three spectra and the blank spectra was subtracted.In order to eliminate the inner filter effects, the fluorescence intensity was corrected using formula (3)[31].

WhereFcorandFobsare the corrected and observed fluorescence intensities, respectively;AexandAemare the absorption of the solution at the excitation and the emission wavelength, respectively.
1.4 Statistical analysis
Three-dimensional modeling was performed using the SWISS-MODEL[32-34]Protein Homology Modeling Servicer by online analysis (http://swissmodel.expasy.org). Subsequent analysis, visualization and preparation of 3D figure were performed using the VMD software.
2 Results and Analysis
2.1 Structural features of SEM revealed by homology modeling
The tertiary structure and topology of SEM, predicted by SWISS-MODEL online tool, was shown in the Fig. 1. Similar to the other members of the staphylococcal enterotoxins-superfamily, SEM adopted the canonical group V superantigen fold. Modeling, based on the crystal structure of template of SEI[35], determined that SEM contained a conserved overall structure made of two major protein domains (Fig. 1). Domain I is an aminoterminal oligosaccharide/oligonucleotide binding (O/B)fold comprised ofβ-barrel including single Cys93residue projected towards the external surface of theβ4-β5 loop.The single free SH-group in Cys93possesses potential for disulfide linkage with another SEM. Domain II is a carboxylterminalβ-grasp domain made of antiparallelβ-strands,which contained the single Trp121and a Zn2+-dependent highaffinity site coordinated by residues His190, His228and Asp230.Furthermore, these two domains are connected by a central,diagonalα-helix. As for the indole ring of the single Trp121,it was located in the external surface of theβ8-strands and surrounded with hydrophilic amino acid residues, indicating that Trp121resides in the relative hydrophobic environment of protein surface. Finally, the predicted content of secondary structure in rSEM, calculated by program STRIDE according to a heuristic algorithm, is described in Table 1.
Fig. 1 Molecular model of monomeric rSEM structure
Table 1 Secondary structure parameters for rSEM predicted from homology modeling and derived from far-UV CD spectra at various temperatures
2.2 Native soluble aggregation and heat-induced expansion of aggregation state of rSEM as monitored by the SDS/native-PAGE
In reducing SDS-PAGE, the purified His-tagged fusion proteins presented the dominant band with molecular weight roughly 30 kDa (Fig. 2A, Lane 1, His-M), which is in agreement with the expected fusion protein molecular weight.Meanwhile, the trace amount of dimer with lower mobility(Fig. 2A, Lane 1, His-D) was also detected, suggesting that His-tagged rSEM possesses the ability to form dimer independent of Histidine-containing peptide. There is a digestion site of thrombin in two integrity structural domains of His-tag and rSEM in fusion protein. After digestion by thrombin and treating with reducingβ-mercaptoethanol in SDS-PAGE, the purified rSEM migrated as a monomer band with a molecular weight of 25 kDa (Fig. 2A, Lane 2 and 4),which correlates well to previously published data[13].However, under non-reducing conditions, only a small amount of monomer (Fig. 2A, Lane 3) with high mobility was observed, the band of dimer (Fig. 2A, Lane 3) at higher molecular weight reigned supreme in SDS-PAGE. Normally,the presence or absence of intermolecular disulfide bonds in a protein could be determined by reducing/non-reducing SDSPAGE. Since disulfide crosslink increases the hydrodynamic radius of SDS-denatured protein, disulfide-bonded species often migrate more slowly in the gel. Therefore, these results correlate well with the homology protein modeling involving in single Cys93, which is inclined to form intermolecular disulfide bond in relative oxidizing condition.
Aggregation of rSEM at room temperature was further analyzed by the native-PAGE, and similar result was observed. Before sample loading, rSEM was treated with SDS instead ofβ-mercaptoethanol to destroy intermolecular non-covalent linkage. Simultaneously, disulfide bridged dimer, which was indeed unfolded by SDS, maintained due to oxidation state. Based on dominant dimer band (Fig. 2C,Lane S, Strip D, close to BSA monomer band with a molecular weight of 68 kDa) presented in the stained gel and together with the results of SDS-PAGE (Fig. 2A, Lane 3), we could draw a conclusion that the dimer of rSEM was formed by intermolecular disulfide bond instead of intermolecular non-covalent linkage. Therefore, the disulfide bond stabilized dimer band in native-PAGE reconfirmed the exposed free SH-group located at single Cys93residue was accessible to another free SH-group of rSEM located at the same position.However, no more other high molecular weight bands would appear in polyacrylamide gel theoretically, since non-covalent aggregation state had been destroyed by SDS-treatment.Contrary to our expectation, the obscure high molecular weight polymer band (HP, much higher than 136 kDa of BSA as a dimer) appeared close to stacking gel (Fig. 2C, Lane S).It was probably due to the heat-generation electrophoretic process created such strong non-covalent aggregates[36-37].
In most conditions for rSEM, just like other SEs[15,18],cryopreservation or evaluation for thermal denaturation of secondary and tertiary structure did in fact cause protein to aggregate, leading to visual precipitation. The manufacturing process of food is commonly using thermal treatment as a major inactivation step for SEs, and hence we set out to clarify the impact of heating on the oligomerization of rSEM.The toxins were exposed to consecutive heating within the temperature interval 30-90 ℃ and were analyzed by SDSPAGE under reducing conditions (Fig. 2B). The consecutive heating was performed in 10 ℃ increments with 10 min equilibration time at each step to ensure thermal increase of the protein samples. This was similar to the temperature increase, while the proteins were exposed to the following CD and fluorescence spectroscopy experiments, with consistent incubation times. With temperature increasing from 30 to 50 ℃, the minor bands of dimer (D) did not have any detectable change. However, at 60 ℃ or above, the aggregation level of dimer increased visibly and reached its maximum at 80 ℃, and then the dimer band disappeared at 90 ℃ just as the fade-out of monomer from 70 to 90 ℃.Accompanied by nearly similar aggregation tendency,tetramer formed at 70 ℃ and mounted maximum at 80 ℃but fade at 90 ℃. Moreover, as seen in Fig. 2B, when heated to 80 ℃, the HP began to form and the monomers decreased in balance with the total amount of loading protein. Besides,the HP displayed at 80 ℃ and the protein aggregation state enhanced so significantly at 90 ℃ that these aggregates failed to penetrate the stacking gel in the SDS-PAGE,indicating that the thermal-induced increase of the extent of protein aggregation was so enormous that aggregates could not be completely dissociated by SDS and reducingβ-mercaptoethanol.
Fig. 2 SDS-PAGE analysis of soluble protein expression, digestion of fusion proteins, aggregation state (A) and consecutive heating of rSEM (B) in absence of Zn2+ and native-PAGE analysis of aggregation state of rSEM (C)in non-reducing loading buffer with SDS at room temperature
Together with above-mentioned results, dimerized rSEM stabilized by disulfide-crosslink is the major aggregate in non-reducing buffer at room temperature. In terms of stable monomeric/dimeric rSEM in reducing/non-reducing conditions respectively, it was obvious that the monomer/dimer of rSEM had no apparent changes below 70 ℃.With the further increase of temperature from 70 to 90 ℃,the grayscale of monomer band gradually reduced in compensation for the development of dimers, tetramers and HP. In brief, the transition of aggregation can be divided into at least three states: 1) From 30 to 60 ℃, rSEM is presented as monomer/dimer in reducing/non-reducing conditions respectively; 2) From 70 to 80 ℃, rSEM formed a series of aggregates, especially tetramer and soluble HP; 3) At 90 ℃or above, the protein aggregation state of rSEM enhanced dramatically, HP dominated in SDS-PAGE.
Previously, increase of protein aggregation state induced by thermal treatment had also been shown for proteins riched inβ-sheets[38-40]. Meanwhile, suchβ-sheets riched proteins would go through secondary structural transitions fromα-helix toβ-sheet during heating and transform from a soluble monomeric state to a cross-β-aggregated state.Therefore, we supposed thatβ-structure riched rSEM would also experiencedα→βtransition accompanied by gradually aggregated with temperature increasing. This supposition could be further validated in the following CD spectra.
2.3 Thermal induced secondary structural changes in rSEM monitored by CD spectra
In order to intensively investigate the stability of secondary structure and structural transitions occurring in heat-treatment, a common method to inactivate toxins during food processing, CD spectroscopy was employed due to its sensitive and direct interpretation of the changes in protein secondary structure[41-43]. As shown in Fig. 3A, the spectrum of rSEM at room temperature (25 ℃) was characterized by two strong CD bands showing negative maxima at 207 nm as well as positive peak at 193 nm and a minor negative shoulder peak around 216 nm. The 193 nm positive band and 207 nm negative band indicated classicalα-helix conformation and the 216 nm negative band demonstrated typicalβ-sheet structure[44]. Furthermore, using DICHROWEB on-line Web server according to section 1.3.5, the CD spectrum was further analyzed to calculate secondary structure contents.As shown in Table 1, the result at room temperature (25 ℃)demonstrated a good correlation between the experimentally determined contents ofα-helical/β-sheeted structure for rSEM and the homology modeling contents according to crystallized SEI[35].
Furthermore, we set out to clarify the impact of heating on the secondary structural elements of rSEM by application of CD spectroscopy. Fig. 3B showed the far-UV spectra of rSEM in intact (dark blue solid dot, 25 ℃) and thermally denatured states, which temperatures were represented by colors of each dot from blue, 30 ℃ to red, 90 ℃. Upon heating, the overall ellipticity in the CD spectra gradually collapsed, indicating the distortion of the secondary structure in rSEM. To further understand the thermal induced structural changes in rSEM, the molar ellipticities at 207 nm in CD spectra were plotted against the temperatures (Fig. 3C),which showed a sigmoidal curve for the conversion of rSEM. Such sigmoidal curve indicates that the temperatureinduced unfolding in terms of secondary structure seemed to occur through a two-state manner, namely from native (N)to “unfolded (U)” states. So the curve can be conveniently divided into three states: 1) The pre-transition state (25-40 ℃),which shows that rSEM exists in a compact native state. 2)The transition state (40-52 ℃), which shows that the rSEM starts to lose its secondary structures and the value varies as unfolding occurs. 3) The post-transition state (above 52 ℃),which shows the denatured state of the “unfolded” rSEM.
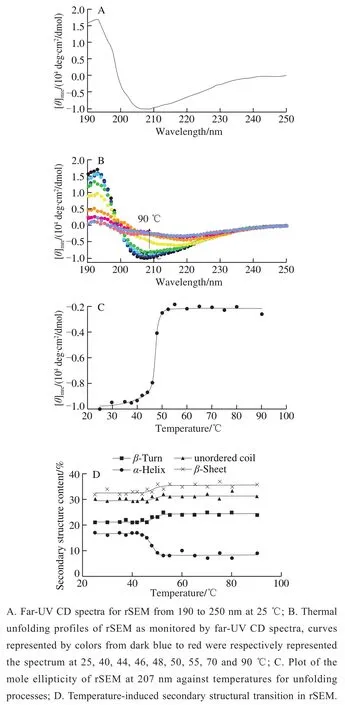
Fig. 3 Circular dichroism (CD) spectra of rSEM exposed to various temperatures
Thermal denaturation data showed that the melting temperature (T1/2), which is defined as the midpoint of the unfolding process and applied to evaluate the thermo-stability of the protein, was 47.3 ℃ for rSEM. This result indicates that rSEM is more sensitive to heat-induced unfolding than other SEs[15,18], which seems to be conflicting with the traditional concept that SEs possess extremely heat-tolerance.Such paradox was partly caused by the reason that it was difficult to directly determine the defined composition of the secondary structure as well as the changes of secondary structure in rSEM upon heating based on the CD spectra only. To throw further light on this paradox, the contents of the secondary structure of rSEM were estimated using DICHROWEB online Web server according to section 1.3.5. As shown in Table 1,all the secondary structures of rSEM were relatively stable below 40 ℃ and the composition of the secondary structures remained almost unchanged. With the further increase of temperature from 42 to 52 ℃,α-helices of rSEM decreased distinctly, whileβ-sheet/turn as well as random coli increased slightly. Noticeably, the decrease ofα-helix accurately corresponded to the increase inβ-sheet/turn and random coli, suggesting thatα→βtransitions induced by heating did happen. While the temperature was above 52 ℃ and finally up to 90 ℃, the contents of the secondary structures of rSEM did not distinctly change and the composition of the secondary structure in so-called “unfolded (U)” state was assigned as 8.2%α-helix, 35.6%β-sheet, 24.4%β-turn and 31.3% unordered structure. According to the classical concept thatβ-structure is much more stable thanα-helices,we suppose that at high temperature rSEM adopts a distinct folded state instead of “unfolded (U)” state and the collapse ofα-helices in central motif does not lead to any obvious change in eitherβ-barrel fold orβ-grasp domain. In brief, the results illustrated that owing to the higher thermal stability forβ-barrel/β-grasp domains than the central, diagonal helix,rSEM is resistant to unfolding at high temperature. Together with the results of aggregation displayed by SDS-PAGE, the aggregation state belonging to state I (30-60 ℃, aggregation remained almost unchanged) was corresponded to the both pre-transition (25-40 ℃) and transition region (40-52 ℃above,α→βtransitions induced by heating) revealed by CD signal. These results suggested that in the range of moderate temperature (40-52 ℃), intramolecularα→βtransition induced by thermal treatment did not immediately result in the formation of a cross-β-aggregated state. In brief,we shall draw a conclusion that such secondary structural transition between 40-52 ℃ lead protein to adopt a new fold withstanding high-temperature treatment. In this regard, our results had never broken the classical concept that SEs had extremely high thermal stability[15,18].
Although CD spectra indicated the transition from native (N)to “unfolded (U)” states of rSEM occurring between 40-52 ℃,the existence of stable intermediate(s) remained uncertain.In order to know whether this distinct folded state is an intermediate state (IS) or the final “unfolded (U)” state upon heating, fluorescence spectroscopic analyses were performed to identify any feasible IS as below.
2.4 Thermal-induced tertiary structural changes in rSEM monitored by the intrinsic fluorescence emission spectroscopy
Although CD spectra indicated that an alternatively folded state with stable secondary structure can be characterized at temperature higher than 55 ℃, the potential heat-induced changes in tertiary structure could not be detected. It is widely accepted that fluorescence measurement directly provides the micro-environmental information around the chromophores in a protein[45]. Thus, the high sensitivity of the fluorescence signal was employed to identify changes of immediate environment of the probe, monitor the structural changes in tertiary structure and especially detect any feasible IS during the heat-induced unfolding process. The main chromophores in rSEM were the single Trp121inβ-grasp fold and the 16 tyrosine residues spreading all over the centralα-helix,β-grasp andβ-barrel fold. The representative fluorescence emission spectra of rSEM were shown in Fig. 4A and Fig. 4B, respectively. At pH 7 and 25 ℃, the fluorescence maxima emission wavelengths of rSEM, with excitation at 278 or 295 nm respectively, were both located around 340 nm (Fig. 4A). The absorption of protein at 278 nm was due to both tyrosine and tryptophan residues. While the emission of tyrosine, which was supposed to occur at 303 nm, had not been observed because of efficiently excited energy transfer from the tyrosine residues to the single Trp121. Such results suggested that aromatic amino acids in recombinant protein spaced favourable Förster distance for resonance energy transfer from tyrosine to tryptophan. Besides, the maximum emission wavelength (λmax)of the single Trp121was located at 340 nm, suggesting that the single Trp121in rSEM was located in relatively hydrophobic environments at room temperature, which is similar to previously published data about tryptophan fluorescence emission maximum in staphylococcal enterotoxin H (SEH)[14].Hence, together with the results revealed by CD and SDSPAGE in the pre-transition region (25-40 ℃), we shall speculate that the rSEM adopts compact native structure in neutral buffer at room temperature. However, at pH 2 and 25 ℃ (Fig. 4B), when excitation at 278 nm, theλmaxof rSEM was blue shifted to 308 nm and the fluorescence intensity was quenched distinctly. Meanwhile, the λmaxof rSEM, excited at295 nm, was located at 352 nm Such discrepancy inλmaxwas due to unfavourable Förster distance for excited energy transfer from tyrosine to tryptophan, incompact folded state of recombinant protein and hydrophilic microenvironment of the single Trp121in acid-unfolded protein. In brief, the 278 nm excitation revealed not only the micro-environment of tryptophan but also the structural compactness of protein. Therefore, the spectra with excitation at 278 nm(λex= 278 nm) at different temperatures were recorded to probe the structural compactness of protein and the changes in micro-environment of tryptophan.
As shown in Fig. 4C, with the increasing of temperature,the intensity of the fluorescence sequentially quenched and theλmaxis preferentially blue shifted at moderate temperature and then red shifted at higher temperature, indicating the structural flexibility of rSEM. To further understand the temperature-induced structural changes in rSEM and whether or not the existence of stable IS upon heating, theλmaxwere plotted against the temperature in Fig. 4D. With the increasing of temperature from 25 to 64 ℃, theλmaxshowed a clear blue-shift from 340 to 331 nm, suggesting that the single Trp121in rSEM was transferred to more hydrophobic environments or partially unfolded conformation led to unfavourable Förster distance for excited energy transfer from tyrosine to tryptophan. With further increase of temperature from 64 to 100 ℃, theλmaxshowed a clear redshift from 331 to 341 nm. Such result clearly indicated that,under high temperature treatment, fluorescence emission maximum seemed identical to compact native state at 25 ℃,demonstrating that, by refolding or something, the microenvironment of the single Trp121was restored or the obstacle for excited energy transfer from tyrosine to tryptophan was eliminated. In any case, the temperature-induced unfolding of rSEM in terms ofλmaxseemed to occur through a multistep process of at least two transitions with a potential IS observed around 64 ℃. However, this fluorescent spectrometric analysis seemed inconsistent with above-mentioned two-state transition revealed by CD measurement.
To clearly monitor theλmaxshifts in the fluorescence emission spectra, the ratio of the fluorescence emission intensity at 350 and 340 nm was measured. As shown in Fig. 4E,the first transition region was observed with a transition midpoint (T1/2) at 47.5 ℃, along with a distinct decrease in the fluorescence intensity ratioF350nm/F340nmand the blueshift of tryptophan fluorescence emission maximum (Fig. 4C and Fig. 4D). This transition region correlated well with the transition curves obtained from CD spectroscopy under same heat-treating conditions (Fig. 3D). Coupled with the apparent decrease inα-helix and slight increase inβ-sheet/turn, the fluorescence emission spectra indicated that the hydrophobicity of micro-environment surrounding Trp121was highly improved or an alternative incompact folded state with unfavourable Förster distance between tyrosine and tryptophan was existed in rSEM at 55 ℃. Besides, the ratioF350nm/F340nmremained unchanged from 55 to 64 ℃, implying that the micro-environment of Trp121or the unfavourable Förster distance between tyrosine and tryptophan was hardly changed within this range. Together with the almost unchanged aggregation state analyzed by SDS-PAGE below 60 ℃, a stable IS was presented around 55-64 ℃. In conclusion, these results clearly demonstrate that such stable IS around 55-64 ℃ has its own characteristics in the high contents ofβ-sheet/turn, compact folded state with flexibile Trp121inβ-grasp domains or incompact folded state with unfavourable Förster distance between tyrosine and tryptophan and stable monomer/dimer in reducing/non-reducing conditions respectively. As a sensitive way to characterize the unfolding IS, the phase diagram was established by plotting the intrinsic fluorescence intensity at 360 nm versus that at 320 nm at all temperatures (Fig. 4F). It could be seen that there were two slope parts in the temperature ranges of 25-60 ℃ and 60-100 ℃ respectively, and where the two lines intersect corresponded to a relatively stable IS compared to native and “unfolded” states upon heating. The overall results demonstrated, once more, that the heat-induced unfolding behavior of rSEM was not a simple two-state model, but a three-state model with a stable IS which was clearly observed around 60 ℃, which was well consistent with the abovementioned stable IS around 55-64 ℃. Plotting theF350nm/F340nmfluorescence intensity ratio with increasing temperature from 52 to 90 ℃, the second transition was observed with aT1/2at 72.5 ℃. Together with unaffected contents of secondary structure detected by CD spectra and the appearance of tetramer or high polymer (HP) as shown by SDS-PAGE from 70 to 80 ℃, all these results clearly indicated that rSEM adopts a folded protein aggregations with high contents ofβ-sheet/turn and quasi-native hydrophobic micro-environment around Trp121even above 80 ℃. Contrary to the popular opinion that protein unfolded and aggregated in the high temperature, rSEM resist to unfold even above 90 ℃ and aggregate to adopt a quasi-native fold instead of the so-called“unfolded (U)” states mentioned in section 2.3. Therefore,by means of spectroscopy and SDS-PAGE measurements upon heating, such quasi-native folded aggregation state with stable secondary/tertiary structure is found and becomes the candidate for heat endurance under high temperature.
Fig. 4 Intrinsic fluorescence emission spectral analysis of rSEM exposed to different temperatures
In conclusion, the most likely explanation for unfolding process of rSEM was that, with temperature increasing from 25-55 ℃, the compact native rSEM became an alternative folded IS around 55-64 ℃. As temperature increased further from 70 to 80 ℃, some intra-molecular regions, like hydrophobic regions, might become exposed to molecular surface and accessible to form new inter-molecular attractive forces through hydrophobic interactions. At 80 ℃ or above,rSEM adopted compact quasi-native folded aggregation state to tolerate high temperature.
It is very interesting that rSEM inβ-grasp domains has such highly dynamic tertiary structure revealed by residue Trp121fluorescent measurement and relative stable secondary structure detected by CD spectra. In order to throw light on such special structural feature involving in rSEM, we employ the well-known principle that change of protein structure would have impact on its biological function. The SEs secreted by the bacteriaStaphylococcus aureus, also be known as superantigen, can elicit massive T cell activation through simultaneous binding to major histocompatibility complex (MHC) class II and T cell receptors. A remarkable character of superantigens, which distinguish them from conventional antigen, is their ability to interact with multiple MHC II alleles. An interesting question is that how to explain the broad MHC cross-reactivity with individual superantigen.Previous crystallographic studies of staphylococcal enterotoxin I (SEI) had shown that staphylococcal superantigens belonging to the zinc family bind to a high affinity site on the class II polymorphicβ-chain[35]. The bridging zinc ion was tetrahedrally coordinated by SEI residues His169, His207and Asp209, which were located on theβ-grasp domain of the C-terminal portion, and by HLADR1 residue His81β, which was located on theα-helix of theβ1 domain ofβ-chain. To account for achieving broad MHC cross-reactivity of SEI, X-ray crystallography study demonstrated above-mentioned zinc-mediated interactions between MHC andβ-grasp domain of superantigen circumvented peptide specificity. Meanwhile, for achieving promiscuous binding to peptides-MHCs, the binding of conserved residues of the polymorphicβ-chain significantly avoided specific interactions with side chains of the antigen peptide. In brief, theβ-grasp motif of the C-terminal portion of SEI, also can be referred to as binding pocket, contacts theα-helix of theβ1 domain of MHC II, as well as the N-terminal portion of MHC-bounded antigen peptide. Therefore, it is reasonable to speculate that the binding pocket in toxin molecule is equipped with significantly structural flexibility to enhance affinity for binding to the diverse MHC alleles, which leads to highly efficient T cell activation, massive release of pyrogenic cytokines and the occurring of toxic shock.
As for SEM and SEI, they all belong to group V superantigen. Hence, we can make reasonable hypothesis that they possess similar superantigen binding model. The binding of SEM to MHC molecule mediated by Zn2+also occurred at the C-terminalβ-grasp domain just like SEI. Therefore,it is most likely that the C-terminalβ-grasp domain is less stable and possibly adopts other folded states to adapt to the variable MHC II molecules. Our spectroscopic measurement demonstrate that the composition ofβ-sheet/turn in rSEM increased slightly from 40 to 52 ℃, which is in contrast to the obvious 9 nm blue-shift of theλmaxand reversed S-shaped curve for fluorescence intensity ratioF350nm/F340nmat the same temperature range. If we suppose that heating process did not expand the size of protein and the Förster distance between tyrosine and tryptophan maintained properly. Abovementioned results demonstrate that the micro-environment of single Trp121, which is located in theβ-grasp domain and close to zinc-mediated MHC II high affinity site, significantly alter upon heating. Therefore, structural flexibility ofβ-grasp domain is obviously detected by fluorescent spectrometry. These results lead to the conclusion that the rSEM possesses well defined regions ofβ-sheet/turn and fluctuating instead of destructive tertiary structure in C-terminalβ-grasp domain. The stable secondary structure is supplied as architecture for rSEM. Moreover, moderate flexibility in C-terminalβ-grasp domain is convenient for induced-fitting MHC II polymorphicβ-chains. Therefore,the fluorescent analysis may partly explain the increase in MHC cross-reactivity with rSEM. In brief, highly dynamic tertiary structure at the C-terminalβ-grasp domain in rSEM can be adapted to significantly structural flexibility to enhance affinity for binding to the diverse MHC alleles.Meanwhile the relative stable secondary structure can provide architectural stability of protein. The result of our experiment is consistent with the traditional inducedfit model of stable integrated architecture for protein and flexible active center of the enzyme for substrate binding.
3 Conclusions
This manuscript may provide the mechanism of thermo-stability and the insight on the unfolding process of rSEM upon heating, which is a common method for food sterilization. rSEM is a two-domain superantigen bridged by a central, diagonal helix. Structural stability analyses of rSEM have been carried out using CD spectra, intrinsic fluorescence spectra and SDS/Native-PAGE during thermal-induced conformational transition. According to above-mentioned results, it was assumed that the process of thermal unfolding of rSEM was involved in a three-state transition model.In brief, there may be three structural entities in rSEM,possibly heat-resistantβ-barrel domain with stable secondary structure, heat-labile central, diagonal helix motif tending to collapse at moderate temperature and heat-stableβ-grasp domain with flexible tertiary structure and stable secondary structure. Although these three distinctive motifs behave completely different upon heating, they unfold cooperatively in their own temperature range. Thus, the thermal induced unfolding behavior of rSEM can be represented by the following scheme:
FS (compact native Folded State, soluble monomer/dimer in reducing/non-reducing buffer, from 25 to 40 ℃) →IS (an alternative folded Intermediate State, soluble monomer/dimer in reducing/non-reducing buffer, from 55 to 64 ℃) → AS (compact quasi-native folded non-covalent Aggregation State, soluble tetramer, soluble HP and even insoluble HPs, above 80 ℃).
Hence, our data support that rSEM could maintain its three-dimensional structure during thermal food processing at neutral pH.
This work describes the conformational and the thermodynamic properties in the unfolding process of rSEM induced by heating, which is the conventional scheme for food disinfection and sterilization. Contrary to our expectation, thermal perturbation does not result in unfolding of the protein with prominent loss of secondary/tertiary structure. At moderate temperature around 55-64 ℃,stable IS was characterized by high contents of secondary structure, well-folded tertiary structure and monomeric/dimeric state. However, such stable IS was prone to aggregate rather unfold with further increase of temperature. While temperature is higher than 90 ℃ for rSEM, stable aggregates with secondary/tertiary structure, which was almost identical to native state except for lower content ofα-helix but higher content ofβ-sheet/turn, were revealed by SDS-PAGE, CD and fluorescence spectroscopy. In conclusion, rSEM clearly aggregate at high temperature, meanwhile it can persist as intact molecules and adopt quasi-native conformation in this temperature range, which can be an argument for its extremely thermo-stability. Thus, this manuscript may provide the insight on the specific thermo-resistant mechanism for rSEM, which is a potential toxin responsible for SFP.
Further work is under way in our laboratory to probe the unfolding process of rSEM induced by pH, chemical denaturants (urea or GdmCl), proteases, molecular dynamic simulation and so on, which may provide more insight on the folding/unfolding mechanism and stable elements for rSEM. These results are helpful for understanding the thermodynamic and kinetic intermediates in its unfolding pathway and identify the important interactions maintaining the native structure of the protein. Therefore, the data would illuminate the specific way to destabilize the rSEM, and thus establish the safer guidelines for food preparation.
